Tadalafil appartiene alla classe degli inibitori selettivi della fosfodiesterasi di tipo 5, con un profilo farmacocinetico caratterizzato da un’emivita terminale di circa diciotto ore. Dopo somministrazione orale viene assorbito rapidamente e raggiunge concentrazioni plasmatiche massime in due ore. La biotrasformazione avviene principalmente tramite CYP3A4 con formazione di metaboliti inattivi, escreti in prevalenza con le feci. L’elevato legame alle proteine plasmatiche (>90%) assicura una distribuzione stabile. Nei confronti delle altre molecole della stessa classe, cialis compresse italia è noto per la durata prolungata dell’attività farmacologica.
No job name
J. Chem. Inf. Comput. Sci. 2000, 40, 773-777 Estimation of Aqueous Solubility for a Diverse Set of Organic Compounds Based on Molecular Topology
Division of Pharmaceutical Chemistry, Department of Pharmacy, POB 56, FIN-00014,
An accurate and generally applicable method for estimating aqueous solubilities for a diverse set of 1297organic compounds based on multilinear regression and artificial neural network modeling was developed. Molecular connectivity, shape, and atom-type electrotopological state (E-state) indices were used as structuralparameters. The data set was divided into a training set of 884 compounds and a randomly chosen test setof 413 compounds. The structural parameters in a 30-12-1 artificial neural network included 24 atom-type E-state indices and six other topological indices, and for the test set, a predictive r2 ) 0.92 and s )0.60 were achieved. With the same parameters the statistics in the multilinear regression were r2 ) 0.88 ands ) 0.71, respectively.
not be representative but compiled from structural analogues. The use of a small and limited set of compounds in the
The aqueous solubility of drug compounds is one of the
training sets leads to models of closed systems, and their
most important factors in determining its biological activity.
general applicability is questionable. This is clearly demon-
In many cases drugs that show a good activity when
strated by the fact that only three of above-mentioned
administered parenterally maybe totally inactive when given
methods6,7,17 have been applied to the test set designed by
orally. In such cases poor oral activity is often due to the
Yalkowsky.19 This test set contains 21 drug molecules and
fact that a sufficient amount of drug to desired response is
environmentally interesting compounds, like pesticides, with
not reached in the site of action. Hence an insufficient
aqueous solubility is likely to hamper bioavailability of the
In our earlier studies we have shown that aqueous
drugs. In recent years high-throughput screening, where
solubilies,17 log S, and partition coefficients,20 log P, for drug
collections of thousands of compounds are screened with
compounds can be estimated with a reasonable accuracy on
the intention of finding relevant biological activity, has
the basis of parameters derived from molecular topology.
proven valuable in finding new lead compounds.1 It has been
In this study we propose a method for estimating log S values
noticed that the synthesis of combinatorial libraries tends to
with the same parameters but for a much larger and diverse
result in compounds with higher molecular weights and
higher lipophilicity, and presumably lower aqueous solubility,than with conventional synthetic strategies. For this reason
computational screens have been suggested and used to selectsublibraries with relevant physicochemical properties to the
The applicability and accuracy of a log S estimation
range of known values, such as lipophilicity and solubility,
method are strongly affected by the size and quality of the
of the orally active drugs.2-5 Hence there is a strong interest
training set used. Experimental aqueous solubility values for
in fast, reliable, and generally applicable structure-based
the compounds used in this study were obtained from the
methods for prediction of aqueous solubility of new drugs
AQUASOL dATAbASE of the University of Arizona21 and
before a promising drug candidate has even been synthesized.
SCR’s PHYSPROP Database.22 A set of 1297 organic
Several approaches have been developed for the prediction
compounds was extracted from these databases and was
of aqueous solubility based on nonexperimental structural
divided into a training set of 884 compounds and a randomly
parameters. These can be divided in substructure (group
chosen test set of 413 compounds. The aqueous solubility
contribution) approaches6-8 and in approaches where pa-
values in 20-25 °C expressed as log S, where S is solubility
rameters are calculated directly from molecular structure,9-18
in mol/L, were used. The log S values of the training set
such as topological indices, molecular volume, molecular
ranged from -11.62 to +1.58 with a mean of -2.70 and
surface area, etc. These methods employ multilinear regres-
standard deviation of 2.01. For the testing set, the smallest
sion or neural network modeling and varying ways of
log S value was -10.41 and the largest +1.13. The mean
structural parametrization. However, currently used methods
and standard deviation were -2.77 and 2.07, respectively.
were developed from relatively small training sets (n ) 200-300). One problem with small training sets is that they might
Three different types of topological indices introduced by
Kier and Hall23-26 were used as structural parameters and
774 J. Chem. Inf. Comput. Sci., Vol. 40, No. 3, 2000
were calculated using the Molconn-Z (Hall Associated
Table 1. Structural Parameters in the Multilinear Regression Model
Consulting, Quincy, MA) software with structure input for
each analyzed compound using the SMILES line notation
code. Simple and valence molecular connectivity indices up
and 1-3 ν), shape indices (1-3κ,
1-3κR), flexibility index (φ), the number of hydrogen-bonding
donors (HBD) and acceptors (HBA), and 39 atom-type
electrotopological state (E-state) indices were calculated.
Cross-correlation analysis showed that pairwise correlations
were r2 < 0.80; hence, all these 55 parameters contain useful
(B) Atom-type Electrotopological State Indicesb
information and could be used in regression analysis.
The multilinear regression (MLR) analysis was performed
with SPSS software (v.8.0, SPSS Inc., Chicago, IL) running
on a Pentium PC. The quality criteria on the fit in MLR
analysis were squared correlation coefficient, r2, standard
deviation, s, and Fischer significant value, F, when all
parameters in the model were significant at the 95%
The artificial neural network simulations were carried out
using NeuDesk software (v 2.20, Neural Computational
sciences, U.K.). A three-layered, fully connected neural
network was trained by the standard back-propagation
learning algorithm with a logistic f(x) ) 1/(1 + e-x)
activation function for both hidden and output nodes. The
same set of parameters as in the MLR equation was tested
in artificial neural networks (ANNs) with one output neuron,
Before the training was started, the input and output values
were scaled between 0.1 and 0.9, and adjustable weights
between neurons were given random values of between -0.5
and 0.5. The learning rate and momentum parameter were
set at 0.1 and 0.9, respectively. The training end point was
determined on the basis of the average training error (E),
Indicator variable for compounds that contain only aliphatic C and
H. b According to Kier and Hall.25 c The number of compounds for
which is the mean-square error between the target and actual
output. The optimal training end point was searched forovertraining the network. It has been accepted that the ratio,
Stepwise and backward methods were employed in the
, of the number of input parameters to the number of
regression analysis, and the following equation containing
weights should be greater than 2.0, although cross-validation
30 parameters was calculated for the training set
allows for the use of smaller values.27,28 Hence networks with8, 10, 12, and 14 neurons in the hidden layer were studied. The network architecture and the training end point giving
the highest coefficient of determination, r2pred, and the loweststandard error s for the predictions of the test set were then
n ) 884, r2 ) 0.89, s ) 0.67, F ) 227.31,
used. To avoid chance effects, the predictions were repeated
10 times with different random starting weights in thenetwork, and the averaged log S values were calculated.
In this equation, n is the number of compounds used in thefit, F is the overall F-statistics for the addition of each
successive term, r2cv is squared correlation coefficient of
In this study the aqueous solubility values of a diverse set
prediction in leave-one-out cross-validation, and ai and Si
of 1297 organic compounds were compiled from two highly
are the regression coefficients and the corresponding struc-
evaluated databases. The data set was divided into a training
tural parameters. The regression coefficients in the equation
set of 884 compounds for developing the MLR and ANN
are indicated in Table 1 with the t-scores of the significant
models and a randomly chosen test set of 413 compounds
parameters, and an example calculation of log S values by
(test set 1) for evaluating the predictive ability of the models.
regression coefficients is given in Table 2. In the leave-one-
Another test set of 21 compounds (test set 2) was also used
out prediction of the MLR model, standard deviation of
and allowed comparison of the predictions with earlier
0.71, is only 0.04 unit higher than for the
fitting model, s ) 0.67. Such a small increase indicates a
Myrdal et al.29 pointed out that the experimental solubility
robustness of the model. Multilinear regression was also able
values can differ by ∼1.0 log unit, especially for compounds
to predict the log S values for 413 compounds in the test set
with a very low log S value. Hence, for the training sets that
with a coefficient of determination of r2
are compiled from relatively complex chemical structures,
standard deviation of prediction s ) 0.71, which are in a
standard deviation, s, will be not lower than ∼0.5 log unit.
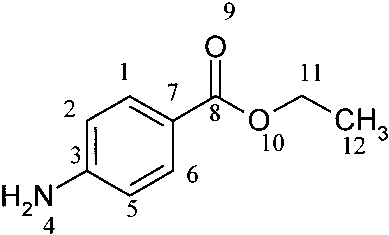
good agreement with the results for the training set. J. Chem. Inf. Comput. Sci., Vol. 40, No. 3, 2000 775 Table 2. E-State Indices Calculated for Benzocaine along with the Atom-type E-State Indicesa and an Example of Calculating log S Valueb by Regression Coefficients Figure 1. Correlation of calculated log S vs observed log S values
for the training set by neural network. a According to Hall and Kier.24 b log S ) -0.4381 + 0.1171 ν -
0.052φ - 0.475HBA - 0.438Ar - 1.96Alif - 0.174SsCH
0.205SssCH2 - 0.08SaaCH + 0.115SdssC - 0.078SaasC + 0.117Ss-NH +
0.048SdO + 0.160SssO - 1.35 ) -1.85 (estimated), -2.32
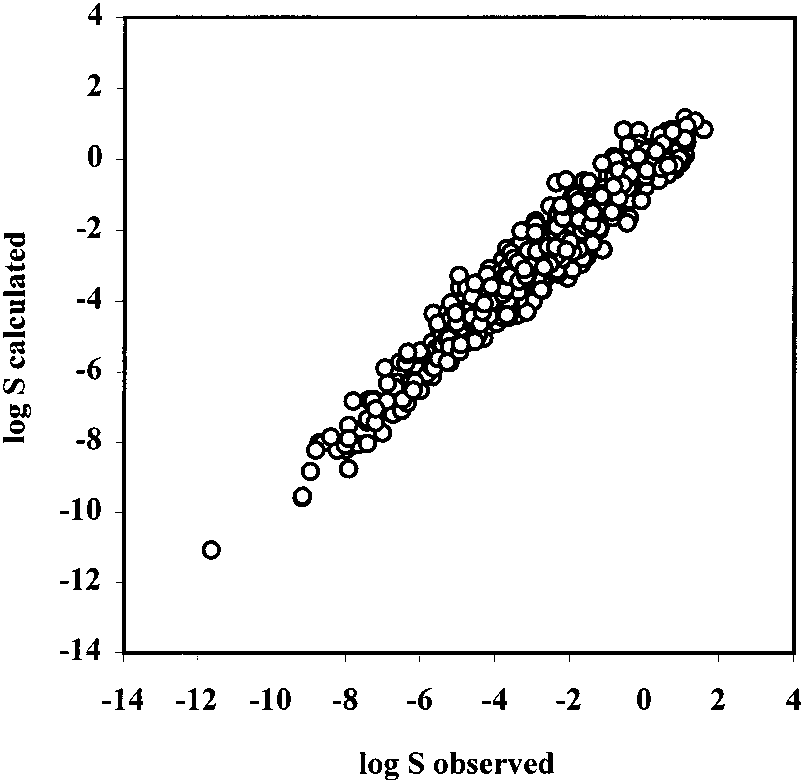
Table 3. Comparison of Predictive Ability of Multilinear Regression and Neural Network Models Using the Same Set of Parameters Figure 2. Correlation of predicted log S vs observed log S values
for the test set 1 by neural network.
training set, r2 ) 0.94, s ) 0.47, and n ) 884 and the test
0.92, s ) 0.60, and n ) 413, respectively.
Statistics for the estimated aqueous solubilities of the
organic compounds in the training set and test sets are
This study. b Our previous study.17
presented in Table 3. The calculated and experimentalaqueous solubilities of the training set and test sets are plotted
It was possible that there were some nonlinear depend-
in Figures 1-3. The list of all compounds and experimental
encies between MLR optimized parameters and log S values.
and estimated log S values is available as Supporting
Hence, an application of nonlinear methods of data analysis
could provide a better modeling of data. The back-propaga-
The general applicability for the prediction ability of
tion artificial neural networks were used to detect the
aqueous solubility was tested by the test set designed by
presence of nonlinear dependencies in the analyzed data set
Yalkowsky.19 This test set is compiled of 21 commonly used
compounds of pharmaceutical and environmental interest.
The same set of the structural parameters as in the
The results of the predictions for this test set are presented
regression equation was used as inputs in neural network
in Table 4. The present multilinear regression and neural
modeling. Several assays were made to find the optimal
network models gave standard deviations s ) 0.88 and 0.63.
training end point and network architecture. The best
In our previous study17 the results by neural network were s
performance of the network was achieved with 12 neurons
) 1.25 for all 21 compounds and s ) 0.55 for a subset of
in the hidden layer with the value of F ) 2.30. The optimal
13 pharmaceuticals. Hence a significant improvement was
training end point, E ) 0.032, required ≈2300 training
achieved, and the predictions were better than those made
epochs when an ANN architecture of 30-12-1 was used.
by Klopman6 and Ku¨hne.7 An interesting point of view is
The neural network was able to estimate, with a reasonable
that Ku¨hne used melting points in their group contribution
degree of accuracy, most of the aqueous solubilities of the
approach and got a better fit for the training set of 694
776 J. Chem. Inf. Comput. Sci., Vol. 40, No. 3, 2000 Table 4. Observed and Predicted Aqueous Solubilities for the Test Set 2 a Outliers in Klopman’s model. b Predicted values not given.
We thank William Howard from Syracuse Research
Corporation for giving the PHYSOPROP database for ouruse and the Technology Development Center in Finland forfinancial support. Supporting Information Available: Appendix I, giving
the names of the compounds used in this study with theircalculated and experimental aqueous solubility values (24pages). This material is available free of charge via theInternet at http://pubs.acs.org.
(1) Gillet, V. J.; Willet, P.; Bradshaw, J. Identification of Biological Active
Profiles Using Substructural Analysis and Genetic Algorithm. J. Chem. Inf. Comput. Sci. 1998, 38, 165-179.
(2) Milne, G. W. A.; Wang, S.; Nicklaus, M. C. Molecular Modeling in
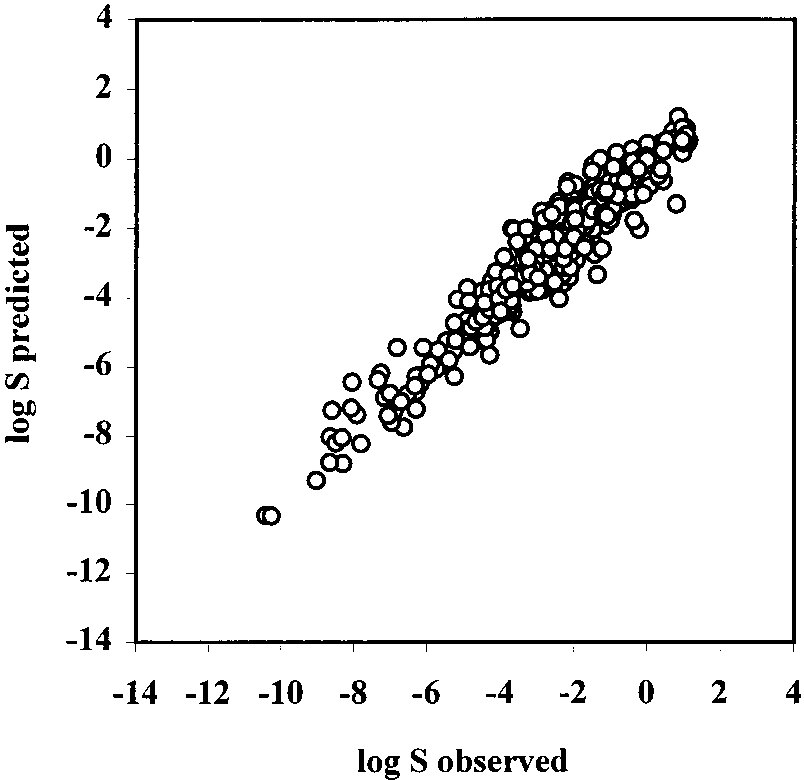
Figure 3. Correlation of predicted log S vs observed log S values
the Discovery of Drug Leads. J. Chem. Inf. Comput. Sci. 1996, 36,
for the test set 2 by multilinear regression (
(3) Ferguson, A. M.; Patterson, D. E.; Garr, C. D.; Underinger, T. L.
Designing Chemical Libraries for Lead Discovery. J. Biomol. Screen.
compounds than Klopman using only group contributors for
1996, 1, 65-73.
a training set of 483 compounds. However, Klopman’s model
(4) Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J.
Experimental and Computational Approaches to Estimate Solubility
(s ) 0.86 and n ) 19) predicted better the solubilities in the
and Permeability in Drug Discovery and Development Settings. AdV.
test set of 21 compounds than Ku¨hne’s model (s ) 1.06 and
Drug DeliVery ReV. 1997, 23, 3-25. n ) 19). Hence we could also ask if the correction term for
(5) Ghose, A. K.; Viswanadhan, V. N.; Wendoloski, J. J. A Knowledge-
Based Approach in Designing Combinatorial or Medical Chemistry
solid compound, melting point, is really necessary for group
Libraries for Drug Discovery. 1. A Qualitative and Quantitative
Characterization of Known Drug Databases. J. Comb. Chem. 1999,
An accurate and generally applicable method for estimat-
ing aqueous solubilities for a diverse set of 1297 organic
(6) Klopman, G.; Wang, S.; Balthasar, D. M. Estimation of Aqueous
Solubility of Organic Molecules by the Group Contribution Approach.
compounds based on multilinear regression and artificial
Application to the Study of Biodegradation. J. Chem. Inf. Comput.
neural network modeling was developed. Topological indices
Sci. 1992, 32, 474-482.
cannot account for three-dimensional and conformational
(7) Ku¨hne, R.; Ebert, R.-U.; Kleint, F.; Scmidt, G.; Schu¨u¨rmann, G. Group
Contribution Methods to Estimate Water Solubility of Organic
effects. Topological indices, however, are attractive because
Chemicals. Chemosphere 1995, 30, 2061-2077.
they can be calculated easily and rapidly and are error-free.
(8) Lee, Y.-H.; Myrdal, P. B.; Yalkowsky, S. H. Aqueous Functional
The results of this study show that a practical solubility-
Group Activity Coefficients (AQUAFAC) 4: Application to Complex
predicting model can be constructed for a large and structur-
Organic Compounds. Chemosphere 1996, 33, 2129-2144.
(9) Nirmalakhandan, N. N.; Speece, R. E. Prediction of Aqueous Solubility
ally diverse set of organic compounds with both multilinear
of Organic Chemicals Based on Molecular Structure. EnViron. Sci.
regression and neural network modeling. Technol. 1988, 22, 328-338. J. Chem. Inf. Comput. Sci., Vol. 40, No. 3, 2000 777
(10) Bodor, N.; Huang, M.-J. Neural Network Studies. 1. Estimation of
(21) Yalkowsky, S. H.; Dannelfelser, R. M. The ARIZONA dATAbASE of
the Aqueous Solubilty of Organic Compounds. J. Am. Chem. Soc.Aqueous Solubility; College of Pharmacy, University of Arizona:
1991, 113, 9480-9483.
(11) Bodor, N.; Huang, M.-J. A New Method for the Estimation of the
(22) Syracuse Research Corporation. Physical/Chemical Property Database
Aqueous Solubility of Organic Compounds. J. Pharm. Sci. 1992, 81, (PHYSOPROP); SRC Environmental Science Center: Syracuse, NY,
(12) Patil, G. S. Prediction of Aqueous Solubility and Octanol-Water
Partition Coefficient for Pesticides Based on Their Molecular Struc-
(23) Kier, L. B.; Hall, L. H. In Molecular ConnectiVity in Structure-ActiVity
tures. J. Hazard. Mater. 1994, 36, 35-43. Analysis; Research Studies Press: Letchworth, 1986.
(13) Nelson, T. M.; Jurs, P. C. Prediction of Aqueous Solubility of Organic
(24) Kier, L. B. Shape Indices of Orders One and Three from Molecular
Compounds. J. Chem. Inf. Comput. Sci. 1994, 34, 601-609.
Graphs. Quant. Struct.-Act. Relat. 1986, 5, 1-7.
(14) Sutter, J. M.; Jurs, P. C. Prediction of Aqueous Solubility for a Diverse
Set of Heteroatom-Containing Organic Compounds. J. Chem. Inf.
(25) Hall, L. H.; Kier, L. B. Electrotopological State Indices for Atoms
Comput. Sci. 1996, 36, 100-107.
Types: A Novel Combination of Electronic, Topological and Valence
(15) Huuskonen, J.; Salo, M.; Taskinen, J. Neural Network Modeling for
State Information. J. Chem. Inf. Comput. Sci. 1995, 35, 1039-1045.
Estimation of the Aqueous Solubility of Structurally Related Drugs.
(26) Hall, L. H.; Story, C. T. Boiling Point and Critical Temperature of a
J. Pharm. Sci. 1997, 86, 450-454.
Heterogeneous Data Set: QSAR with Atom Type Electrotopological
(16) Huibers, P. D. T.; Katritzky, A. R. Correlation of the Aqueous
State Indices Using Artificial Neural Networks. J. Chem. Inf. Comput.
Solubility of Hydrocarbons and Halogenated Hydrocarbons with
Sci. 1996, 36, 1004-1014.
Molecular Structure. J. Chem. Inf. Comput. Sci. 1998, 38, 283-292.
(27) Manallack, D. T.; Livingstone, D. J. Artificial Neural Networks:
(17) Huuskonen, J.; Salo, M.; Taskinen, J. Aqueous Solubility Prediction
Application and Chance Effects for QSAR Data Analysis. Med. Chem.
of Drugs Based on Molecular Topology and Neural Network Model-
ReV. 1992, 2, 181-190.
ing. J. Chem. Inf. Comput. Sci. 1998, 38, 450-456.
(18) Mitchell, B. E.; Jurs, P. C. Prediction of Aqueous Solubility of Organic
(28) Andrea, T. A.; Kalayeh, H. Application of Neural Notworks in
Compounds from Molecular Structure. J. Chem. Inf. Comput. Sci.
Quantitative Structure Activity Relationships of Dihydrofolate Re-
1998, 38, 489-496.
ductase Inhibitors. J. Med. Chem. 1991, 34, 2824-2836.
(19) Yalkowsky, S. H.; Banerjee, S. In Aqueous Solubility. Methods of
(29) Myrdal, P. B.; Manka, A. M.; Yalkowsky, S. H. AQUAFAC 3:
Estimation for Organic Compounds; Marcel Dekker: New York, 1992.
Aqueous Functional Group Activity Coefficients: Application to the
(20) Huuskonen, J. J.; Villa, A. E. P.; Tetko, I. V. Prediction of Partition
Estimation of Aqueous Solubility. Chemosphere 1995, 30, 1619-1637.
Coefficients Based on Atom-Type Electrotopological State Indices. J. Pharm. Sci. 1999, 88, 229-233.
http://web.lexis-nexis.com/universe/printdoc LexisNexis™ Academic Copyright 2003 The New York Times Company The New York Times June 29, 2003, Sunday, Late Edition - Final SECTION: Section 3; Page 1; Column 2; Money and Business/Financial Desk LENGTH: 2846 words HEADLINE: Who's Minding the Drugstore? BYLINE: By MELODY PETERSEN BODY: FEDERAL regulators decided last y
The Philosophy of Mediation: Questions and Responses from Diverse Parts of the World and Especially from the Islamic Tradition The 3rd European Conference on Mediation, “ Mediation and Civil Society in Europe – Towards a new mindset ”, Bourg la Reine, France, May, 2010. Abstract: In this paper, the author highlights the fact that the concept of mediation exists in many cultures and
 J. Chem. Inf. Comput. Sci., Vol. 40, No. 3, 2000 775
J. Chem. Inf. Comput. Sci., Vol. 40, No. 3, 2000 775 776 J. Chem. Inf. Comput. Sci., Vol. 40, No. 3, 2000
776 J. Chem. Inf. Comput. Sci., Vol. 40, No. 3, 2000