Tadalafil appartiene alla classe degli inibitori selettivi della fosfodiesterasi di tipo 5, con un profilo farmacocinetico caratterizzato da un’emivita terminale di circa diciotto ore. Dopo somministrazione orale viene assorbito rapidamente e raggiunge concentrazioni plasmatiche massime in due ore. La biotrasformazione avviene principalmente tramite CYP3A4 con formazione di metaboliti inattivi, escreti in prevalenza con le feci. L’elevato legame alle proteine plasmatiche (>90%) assicura una distribuzione stabile. Nei confronti delle altre molecole della stessa classe, cialis compresse italia è noto per la durata prolungata dell’attività farmacologica.
Pii: s0891-5849(99)00247-6
Free Radical Biology & Medicine, Vol. 28, No. 3, pp. 351–360, 2000
Copyright 2000 Elsevier Science Inc. PII S0891-5849(99)00247-6 Original Contribution
INCREASED LIPOPROTEIN OXIDATION IN ALZHEIMER’S DISEASE
SVEN SCHIPPLING,* ANATOL KONTUSH,* S¨ONKE ARLT,* CARSTEN BUHMANN,† HANS-J¨ORG ST¨URENBURG,†
ULRIKE MANN,‡ TOMAS M¨ULLER-THOMSEN,‡ and ULRIKE BEISIEGEL*
*Medical Clinic, †Neurological Clinic, and ‡Psychiatric Clinic, University Hospital Hamburg, Hamburg, Germany;
(Received 19 October 1999; Accepted 16 November 1999)
Abstract—Oxidation has been proposed to be an important factor in the pathogenesis of Alzheimer’s disease (AD) and amyloid  is considered to induce oxidation. In biological fluids, including cerebrospinal fluid (CSF), amyloid  is found complexed to lipoproteins. On the basis of these observations, we investigated the potential role of lipoprotein oxidation in the pathology of AD. Lipoprotein oxidizability was measured in vitro in CSF and plasma from 29 AD patients and found to be significantly increased in comparison to 29 nondemented controls. The levels of the hydrophilic antioxidant ascorbate were significantly lower in CSF and plasma from AD patients. In plasma, ␣-carotene was significantly lower in AD patients compared to controls while ␣-tocopherol levels were indistinguishable between patients and controls. In CSF, a nonsignificant trend to lower ␣-tocopherol levels among AD patients was found. Polyunsaturated fatty acids, the lipid substrate for oxidation, were significantly lower in the CSF of AD patients. Our findings suggest that (i) lipoprotein oxidation may be important in the development of AD and (ii) the in vitro measurement of lipid peroxidation in CSF might become a useful additional marker for diagnosis of AD. Keywords—Alzheimer’s disease, Antioxidants, Cerebrospinal fluid, Free radicals, Lipid peroxidation, Lipoproteins, Plasma INTRODUCTION
[8,9]. Evidence for increased oxidative damage in ADincludes studies showing that brain tissue from AD pa-
Alzheimer’s disease (AD) is a demential disorder with
tients has higher levels of oxidized proteins [3], ad-
increasing prevalence in the elderly population in the
vanced glycation end products [10,11], and 4-hy-
Western world. Senile plaques and neurofibrillary tan-
droxynonenal-derived adducts [12,13] than tissue from
gles are salient features in AD brains at autopsy and the
nondemented elderly controls. Lipid peroxidation is in-
histopathological hallmarks of clinical dementia. Apart
creased in the brain in AD [14] and the transition metal
from rare cases of early onset AD with causative muta-
ions Cu(II) and Fe(III), capable of producing reactive
tions in the amyloid precursor protein (APP) or preseni-
oxygen species, have been shown to be elevated in AD
lin (PS 1 and PS2) genes [1], the etiology of AD is
brain tissue [15,16]. In addition, APP can reduce Cu(II)
multifactorial [2]. In the framework of such a concept,
to Cu(I), a highly reactive form [17].
several authors have proposed a pivotal role for oxida-
The oxidation hypothesis is supported by the initial
tion in the pathogenesis of AD in recent years [3–7]. The
large clinical trial that proposed a beneficial effect of
central nervous system is especially vulnerable to oxida-
␣-tocopherol and selegiline by slowing the progression
tive stress as a result of the brain’s high oxygen con-
of the disease [18]. Alpha-tocopherol is also known to be
sumption, abundant lipid content, and relative paucity of
effective against lipid peroxidation and to reduce the
antioxidant compounds compared with other tissues
neurotoxicity of amyloid  (A), a major component ofsenile plaques [19]. The exact mechanisms responsible
Address correspondence to: Dr. Anatol Kontush, Biochemisches
for increased oxidation in AD brain remain unclear and
Labor, Pav. 39, Medizinische Kern- und Poliklinik, Universita¨tskran-
there is not yet enough evidence to decide whether oxi-
kenhaus Eppendorf, Martinistraße 52, 20246 Hamburg, Germany;
dation is a primary phenomenon inducing neurodegen-
Tel: ϩ49 (40) 42803-4449; Fax: ϩ49 (40) 42803-4592; E-Mail:[email protected].
eration or is secondary to cell death and loss of neurons.
A has been implicated as an oxidant involved in
Sample collection and preservation
the pathogenesis of AD [5,6,20]. Two facts make Apotentially important for lipid peroxidation: soluble
From each patient, 1 ml of CSF (obtained as a
A is found in biological fluids like cerebrospinal
surplus of diagnostic lumbar puncture) and 10 ml of
fluid (CSF), which is in direct contact with the brain
ethylenediaminetetraacetic acid (EDTA)-anticoagu-
[21], and in both CSF [22] and plasma [23], A is
lated blood were sampled at the same visit and imme-
diately placed on ice. Blood was centrifuged at 4°C for
Lipoproteins in the density range of plasma high-
10 min at 2500 rpm to obtain plasma and the cellular
density lipoproteins (HDL) have been found in CSF
buffy coat for DNA preparation. CSF and plasma were
[24,25]. They contain polyunsaturated fatty acids
freshly frozen under argon or nitrogen at Ϫ80°C, not
(PUFA), the major substrate for lipid peroxidation, as
later than 30 min after puncture. The buffy coat was
well as lipophilic antioxidants such as tocopherols.
stored at Ϫ20°C. Samples were not stored longer than
Plasma lipoproteins have been shown to be highly sus-
3 months. The samples were thawed at room temper-
ceptible to oxidative modifications [26], a mechanism
playing a crucial role in the pathogenesis of atheroscle-rosis. Changes in the chemical composition of CSF li-
poproteins have been recently reported in AD [27]. Werecently found that lipoproteins of human CSF are easily
Oxidation of CSF and plasma was monitored as a
oxidized in vitro [28]. The increased oxidation damage in
change in the sample absorbance at 234 nm. This param-
the brain of AD patients led us to believe that lipoprotein
eter has been shown to reflect the level of lipid hydroper-
oxidation could be important in AD as it is in athero-
oxides in isolated low-density lipoprotein (LDL) oxi-
sclerosis. To assess this hypothesis, we analyzed plasma
dized under in vitro conditions [30]. Lipid hydroxides are
and CSF samples from 29 AD patients and 29 nonde-
other products of LDL oxidation that have conjugated
mented controls for their lipid content, the amount of
diene structure and specifically absorb at 234 nm. How-
lipophilic and hydrophilic antioxidants, and the oxidiz-
ever, they comprise only a small percentage of hydroper-
ability of the samples in vitro. We found that CSF
oxides formed during lipoprotein oxidation [31]. When
lipoproteins in AD patients were more susceptible to
lipoproteins are oxidized in diluted plasma or CSF,
oxidation than those from nondemented controls. The
changes in the absorbance at 234 nm correlate with other
increased lipoprotein oxidation in CSF can provide a
indices of lipid peroxidation, such as consumption of
useful additional marker in the clinical diagnosis of AD
PUFAs and accumulation of cholesterol linoleate hy-
and in particular it might allow a verification of the
droperoxide [28,32]. Furthermore, when oxidized plasma
was treated with sodium borohydride, to eliminate hy-droperoxides, and then extracted with hexane, no in-crease in the absorbance of the extract at 234 nm was
MATERIALS AND METHODS
found in comparison with unoxidized samples (data not
shown). These data justify the use of absorbance at 234nm as a specific measure for the accumulation of lipid
AD patients (n ϭ 29) and control subjects (n ϭ 29)
were recruited in the psychiatric clinic and the neurolog-
To register oxidation kinetics, CSF was diluted 10-
ical clinic of Hamburg University Hospital. The AD
fold with phosphate-buffered saline (PBS), containing
patients were all seen in the outpatient “memory clinic”
0.6 M NaCl, pH 7.4, treated with Chelex 100 ion-ex-
and diagnosed as “clinically probable” according to the
change resin (Bio-Rad, Munich, Germany) for 1 h to
NINCDS-ADRDA and DSM-IV criteria for primary de-
remove transition metal ions. The samples were oxidized
generative dementia, Alzheimer type [29]. All AD pa-
at 37°C either in the absence (autoxidation condition) or
tients were in an early stage of the disease, mobile, in a
in the presence of the exogenous oxidant 2,2Ј-azobis-(2-
good general nutritional state, and did not take antioxi-
amidinopropane) hydrochloride (AAPH; Polysciences,
dant supplements. The control subjects attended the neu-
Inc., Warrington, PA, USA) at 100 M. The absorbance
rological clinic and underwent lumbar puncture for di-
was continuously registered spectrophotometrically at 5
agnostic purpose. Patients with degenerative disorders
min intervals over 50 h at 37°C in quartz cuvettes tightly
were excluded, as were all patients with clinically evi-
sealed with Nescofilm to prevent evaporation.
dent cognitive impairment. Informed consent according
Plasma was diluted 150-fold with PBS and incubated
to the declaration of Helsinki was obtained before lum-
at 37°C for 20 h in the absence of exogenous oxidants
bar puncture and the study was approved by the Ethical
(autoxidation condition) or in the presence of AAPH
(330 M) [33]. The absorbance was measured at 234 nm
as described for CSF and the formation of conjugated
(UV) detection at 267 nm [36]. One hundred microliters
dienes was quantified by the mean oxidation rate during
CSF were diluted 1:1 with 10% meta-phosphoric acid
and centrifuged for 3 min at 13,000 rpm and 4°C. Onehundred microliters of the supernatant were injected intothe HPLC system using a solution of 0.1 M Na HPO ,
2.5 mM EDTA, and 2.0 mM tetrahexyl ammonium chlo-
CSF and plasma fatty acids and CSF cholesterol were
ride, pH 3.0, as a mobile phase, running at 1.0 ml/min.
measured by capillary gas chromatography with flame
Plasma ascorbate was measured photometrically as de-
ionization detection [34]. One hundred microliters of
CSF were mixed with 2 ml chloroform/methanol (2:1vol/vol), and 100 l of heptadecanoic acid (200 mg/l)
and 25 l 5␣-cholestane (100 mg/l) were added as in-ternal standards; 25 l butylated hydroxytoluene (BHT,
The apolipoprotein (Apo) E genotype was determined
0.2 M) was added as antioxidant. The chloroform extract
using the restriction isotyping method as described else-
was evaporated under nitrogen, the dried lipids were
dissolved in 250 l toluene, and fatty acids were deri-vatized with 500 l of 0.5 M anhydrous sodium methox-
ide for 15 min at 50°C. The mixture was neutralized with1 ml 2.5% acetic acid and extracted with 250 l hexane.
Between-group differences in continuous variables were
The supernatant was evaporated under nitrogen and 100
analyzed by Student’s t-test for independent groups. Differ-
l dimethylformamide were added. Cholesterol was si-
ences in dichotomous variables were analyzed by Fisher’s
lylated by incubation with N,O-bis(trimethylsilyl)triflu-
exact test. Pearson’s moment-product correlation coeffi-
oroacetamide for 30 min at room temperature. After a
cients were calculated to evaluate relationships between
final evaporation, the pellet was dissolved in 20 l tol-
variables. Multiple regression was performed on oxidation
uene, 2 l of which were injected into a Hewlett-Packard
parameters to elucidate the extent to which the oxidizability
5890 Series II gas chromatograph (Hewlett Packard, Palo
was specifically influenced by the presence of the disease,
Alto, CA, USA). CSF saturated fatty acids (SFA) were
rather than by other independent factors. All results are
calculated as the sum of palmitic, stearic, and arachidic
expressed as means Ϯ standard deviations. The quality of
acids; monounsaturated fatty acid (MUFA) was defined
the assays was controlled by measuring the assay variabil-
as oleic acid; and PUFA was defined as the sum of
ity, which was not higher than 8% for all the parameters
Plasma fatty acids were measured by capillary gas
chromatography with flame ionization detection as de-
scribed elsewhere [34]. Plasma cholesterol and triglyc-erides were quantified by commercially available enzy-
matic kits (Boehringer Mannheim, Mannheim, Germany).
Clinical data of 29 AD patient and 29 controls are
given in Table 1. The AD patients had a mean Mini
Mental Status Examination score of 19 Ϯ 5. Due to thesignificantly higher mean age of the AD patients (72 vs.
Alpha-tocopherol, ␣- and -carotene, ubiquinol-10,
55 years), we performed a subgroup analysis including
and ubiquinone-10 were measured as the main lipophilic
only individuals Ն 60 years to eliminate a possible
antioxidants in plasma. For CSF, only the levels of
age-related influence on the data. Twenty-six patients
␣-tocopherol and -carotene were determined. The an-
remained in the AD group and 14 in the age-matched
tioxidant content was quantified by reversed-phase high-
control subgroup. All data were analyzed for the total
performance liquid chromatography (HPLC) with elec-
group and the individual subgroups. The mean age of
trochemical detection as described elsewhere [35],
onset was 68 years in the total AD group and 70 years in
except that the system was calibrated using an external
the AD subgroup, and the mean time since diagnosis of
the disease was 3.6 years in both groups.
As expected, the frequency of the 4 allele of Apo
E was significantly higher in the AD patients, .36 vs. .07 in controls. This frequency is in good accordance
Ascorbate, the major hydrophilic antioxidant in CSF,
with epidemiological data [39]. The between-group
was measured by reversed-phase HPLC with ultraviolet
comparison revealed a significantly higher rate of cur-
* p Ͻ .01, † p Ͻ .05 vs. corresponding control group.
rent smokers among the controls. No other parameter
ical fluids, in which lipids are present as lipoproteins. We
of potential interest in the context of oxidative stress,
previously developed a method to record lipoprotein
such as presence of coronary heart disease (CHD),
oxidation in human plasma via photometric measure-
hypertension and diabetes or plasma lipids showed a
ment of the kinetic of conjugated diene formation [33], a
significant between-group difference (Table 1).
protocol that was subsequently adapted for the charac-terization of lipoprotein oxidation in CSF [28].
A typical time course of the absorbance at 234 nm
exhibited three consecutive phases: the lag phase, during
Given the difficulties in measuring oxidative stress in
which the oxidation rate was close to zero; the propaga-
vivo, we decided to assess lipid peroxidation in biolog-
tion phase, representing rapid accumulation of lipid per-oxides; and the plateau phase with an oxidation rate closeto zero again (Fig. 1A). The absorption of oxidizing CSFwas found to parallel the time course of phosphatidyl-choline hydroperoxides accumulation measured byHPLC with UV detection (Fig. 1B) and was correlatedwith the consumption of antioxidants in the sample [28].
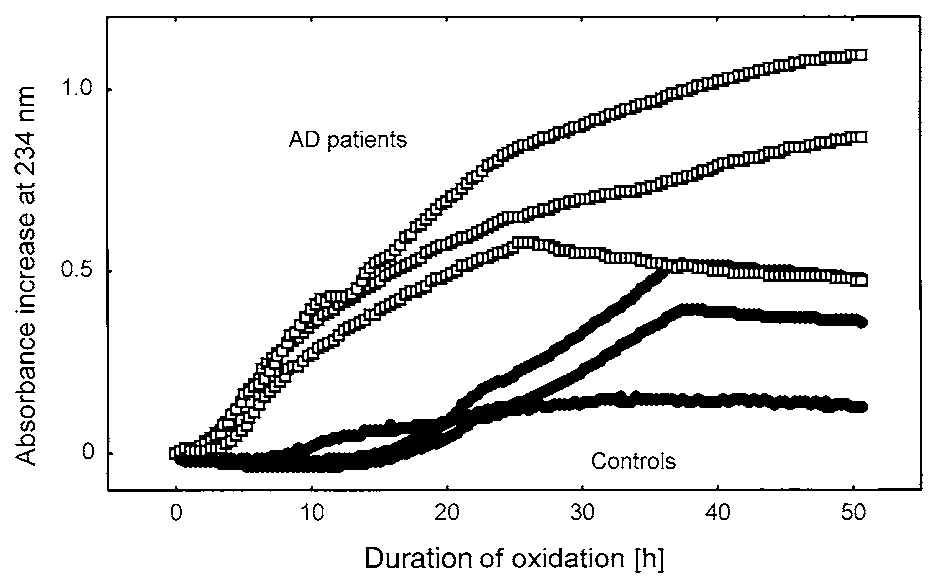
Typical oxidation kinetics from three AD patients and
three controls are shown in Fig. 2. AD patients exhibitedan increased oxidation rate in the lag phase and a clearlyshorter lag phase duration, indicating a more rapid oxi-dation of CSF lipoprotein in AD patients compared withcontrols. To compare the kinetics for all subjects, themean oxidation rate during the initial phase and the lag
Fig. 1. (A) Typical absorbance increase of CSF at 234 nm during itsautoxidation. Time points were taken every 5 min. CSF was diluted10-fold and incubated at 37°C in the sealed cuvettes. (B) Lag, propa-gation, and plateau phases of the oxidation. As in (A), but the accu-
Fig. 2. Typical absorption increase at 234 nm of CSF obtained from
mulation of lipid hydroperoxides was measured by absorption increase
three representative AD patients (ᮀ) and three representative control
at 234 nm (ᮀ) and HPLC (■); fewer time points were sampled, as
subjects (●). CSF was diluted 10-fold and incubated at 37°C in the
absence of exogenous oxidants to determine autoxidation.
Fig. 3. (A) Rate and (B) lag phase of autoxidation of CSF obtained from AD patients and control subjects. CSF was diluted 10-foldwith PBS and incubated at 37°C in the absence of exogenous oxidants. Bars correspond to the mean values calculated for each group. Each open circle (E) and open square (ᮀ) corresponds to one subject older and one subject younger than 60 years, respectively. Significance of the difference between the groups is shown as a p value.
phase duration were calculated for each curve. When
There was no significant difference between the CSF
both parameters were compared between all AD patients
samples of patients with AD and controls in the levels of
and all controls, the oxidizability of CSF from patients
total protein, cholesterol, total fatty acids (TFA), and
with AD was found to be significantly higher. The mean
MUFA (Table 2). These data indicate that differences
oxidation rate during the initial phase expressed as nano-
observed in other parameters cannot be due to differ-
moles of dienes liter per minute (nM/min), was signifi-
ences in CSF volume. The relative level of PUFA in CSF
cantly higher (p ϭ .0008; Fig. 3A) and the duration of the
(expressed as a percentage of TFA) was significantly
lag phase was significantly shorter (p ϭ .00003; Fig. 3B)
lower in the patients with AD (p ϭ .001), whereas SFA
in the AD group. The age of the patients had no influence
were found to be relatively increased (p ϭ .02; Table 2).
on the sample oxidizability, so that the difference re-
There was a negative correlation between the relative
mained similarly significant in the age-matched sub-
PUFA content and both the autoxidation rate and the
groups. The differences were similarly pronounced when
oxidation rate with AAPH (r ϭ Ϫ0.29, p ϭ .03; r ϭ
AAPH, a chemical initiator of the oxidation, was added
Ϫ0.43, p ϭ .001, respectively).
To complement these methods, the levels of hydro-
Table 2. Protein, Lipids, and Antioxidants in CSF of AD Patients and Control Subjects
a Weight percentage of TFA. * p Ͻ .001; † p Ͻ .01; ‡ p Ͻ .05 vs. corresponding control group.
philic and lipophilic antioxidants were measured in the
in patients without AD [33]. When the mean initial
same samples (Table 2). Ascorbate, the major hydro-
oxidation rates were compared, the rates in AD plasma
philic antioxidant in CSF [40], was significantly lower in
samples were significantly higher. This was the case for
the both the total group and the age-matched AD sub-
both autoxidation and oxidation by AAPH, which in-
group compared to controls (p ϭ .006 and .007, respec-
creased the oxidation rates in both groups in parallel
tively). Additionally, as expected, ascorbate levels were
negatively correlated with CSF autoxidation rate (r ϭ
When plasma lipids of patients with AD and controls
Ϫ0.32, p ϭ .02). Alpha-tocopherol, the major lipophilic
were compared, neither triglycerides and total choles-
antioxidant in lipoproteins, tended to decrease in AD
terol (Table 1), nor total fatty acids, saturated, monoun-
patients’ CSF but this difference did not reach statistical
saturated, and polyunsaturated fatty acids (data not
significance. The difference in -carotene values was
The levels of antioxidants in plasma were in good
accordance with the results of the oxidation kinetics
(Table 3). In the total AD group, the hydrophilic antiox-
The plasma oxidation kinetics observed in the present
idant ascorbate was significantly decreased (p ϭ .02), but
study were in accordance with previously reported data
was only slightly lower in the age-matched AD subgroup
Fig. 4. Initial oxidation rate of plasma obtained from AD patients and control subjects. Plasma was diluted 150-fold with PBS andincubated at 37°C in the absence of exogenous oxidants (autoxidation) and in the presence of AAPH (330 M). Bars correspond withthe mean values calculated for each group. Each open circle (E) and open square (ᮀ) corresponds to one subject older and one subjectyounger than 60 years, respectively. Significance of the difference between the groups is shown as a p value.
Table 3. Antioxidants in Plasma of AD Patients and Control Subjects
* p Ͻ .001; † p Ͻ .05 vs. corresponding control group.
(p ϭ .40). The only lipophilic antioxidant that showed
DISCUSSION
significant difference between the groups was ␣-carotene(p ϭ .00001). There were no differences in the levels of
Our study revealed a highly significant increase in
␣-tocopherol, -carotene (Table 3), ubiquinol-10, and
lipoprotein oxidizability in vitro for both CSF and
ubiquinone-10 (data not shown). These results were con-
plasma samples from AD patients compared with con-
sistent with the age-matched analysis (Table 3) and the
trols. In parallel, a decrease in antioxidant levels in both
lipid-normalized values expressed as picomoles per mil-
biological fluids was demonstrated. These data support
ligram (pmol/mg) total cholesterol and triglycerides
the concept of oxidation as an important factor in the
(data not shown). The latter can be explained by the
pathogenesis of AD and might provide a useful addi-
comparable lipid levels in both groups. The plasma au-
tional tool in the diagnosis of the disease.
toxidation rate was negatively correlated with the content
Oxidation of plasma lipoproteins has been studied
of ascorbate (r ϭ Ϫ0.29, p ϭ .05) and with the lipophilic
quite extensively [26,41] and data obtained in plasma
antioxidants ␣-tocopherol and ␣-carotene (r ϭ Ϫ0.37,
samples of patients with CHD and hyperlipidemia versus
Ϫ0.73; p ϭ .01, Ͻ .001, respectively), as was plasma
controls demonstrated differences between these groups
oxidation rate by AAPH with the levels of ascorbate and
[33]. Human CSF contains lipoproteins with properties
␣-carotene (r ϭ Ϫ0.30 and Ϫ0.51; p ϭ .04 and Ͻ .001,
similar to those of HDL from blood plasma [24,25,42].
CSF lipoproteins have, however, not yet been fully char-acterized, and no data on their oxidation are available. We have recently shown that lipoproteins of human CSF
Correlations and results of multiple regression
are oxidatively modified during CSF incubation at 37°C[28]. Our present data show reproducible differences
To elucidate a potential relationship between the ex-
between AD patients and controls in the time course of
tent of oxidizability of CSF and plasma, we examined the
CSF oxidation in vitro. In addition, CSF PUFA, the main
correlation between CSF and plasma oxidizability pa-
substrate for lipid peroxidation, was found relatively
rameters. The autoxidation rate in CSF correlated posi-
reduced in AD, which was in accordance with data
tively with plasma oxidation rates under both oxidizing
published by others [27]. While the increased formation
conditions (autoxidation: r ϭ 0.56, p Ͻ .001 and oxida-
of conjugated dienes can also be shown in plasma, the
tion by AAPH: r ϭ 0.49, p Ͻ .001). CSF lag phase
relative reduction of PUFA cannot be demonstrated in
duration also showed a negative correlation with plasma
plasma samples. This observation can be due to the much
autoxidation rate (r ϭ 0.45, p ϭ .001).
higher amount of fatty acids in plasma.
To detect a potential influence of factors other than
It has been previously shown that the oxidizability of
AD on the oxidation kinetics, multiple regression was
plasma LDL is determined by three major factors: (i)
performed on oxidation parameters as dependent vari-
levels of antioxidants; (ii) levels of substrate for oxida-
ables, using the categoric variables AD, sex, Apo E 4
tion, such as PUFA or other lipids; and (iii) levels of
allele, current smoking, increased alcohol intake, pres-
preformed oxidation products or other substances able to
ence of CHD and hypertension, and continuous vari-
accelerate LDL oxidation [43,44]. Increased oxidizabil-
able age as independent variables. The presence of AD
ity of plasma and CSF lipoproteins in AD might there-
significantly influenced every oxidation parameter in
fore be theoretically related to (i) lower levels of anti-
CSF and plasma. From the other parameters only sex
oxidants, (ii) higher levels of substrate for oxidation, and
entered the final regression equation once. The rela-
(iii) presence of (yet unidentified) oxidants. We did find
tively small number of patients did not, however,
lower levels of antioxidants in CSF and plasma from AD
allow a correlation between the severity of disease and
patients. However, the CSF level of oxidation substrate
(PUFA) was significantly lower in AD. This finding
could be due to elevated oxidation of CSF lipids in AD
in accordance with recent studies showing that AD brain
in vivo, i.e., to higher “preoxidation” of CSF samples
tissue has higher amounts of oxidatively modified bi-
used to measure the oxidizability. This phenomenon
omolecules, as well as higher basal levels of lipid per-
might be associated, in turn, with elevated levels of
oxidation, than tissue from control subjects [3,10 –14].
oxidation products, or other substances able to accelerate
Alternatively, low levels of antioxidants in AD may
in vitro oxidation, in CSF from AD patients. The finding
result from insufficient dietary supply. However, this is
that CSF oxidizability was increased in AD despite lower
unlikely to be the case in our study, since a subset of 10
PUFA levels, might therefore be explained by increased
AD patients showed normal plasma levels of vitamin B12
levels of these, yet unidentified, oxidants.
and normal body mass index values (data not shown). In
We have previously reported that diluted CSF can be
addition, we recruited AD patients who were in the early
oxidized without adding exogenous oxidants, i.e., auto-
stage of disease, as indicated by the short time since its
catalytically [28]. The autoxidation of CSF was com-
diagnosis. This suggests the patients with AD had a
pletely inhibited by EDTA, indicating that it was cata-
lyzed by transition metal ions, such as Cu(II) and/or
Because APP, A, and senile plaques, the main mark-
Fe(III). These ions are present in native CSF as redox-
ers for AD, have been shown to cause increased oxida-
inactive complexes with the metal-binding proteins cer-
tive stress [5,6,17,56], oxidation seems to be secondary
uloplasmin and transferrin [45] but can be released, when
to the formation of A and senile plaques, thus promot-
intact protein structure is disturbed under some patho-
ing the course of the disease rather than being causal.
logic conditions, which may occur in vivo [46,47]. Pro-longed incubation in vitro at 37°C is likely to disturb the
Oxidation can induce protein dysfunction and cell death,
structure of metal-binding proteins, enabling release of
and neuronal cell death contributes to further oxidation.
Cu(II) and Fe(III) in a catalytically active form. Cu(II)
Therefore, oxidation might be both a cause and conse-
and Fe(III) [15,16] as well as their carriers ceruloplas-
quence of the degenerative processes in AD brains.
min, transferrin, and p97 [45,48-50] have all been shown
The correlation found between plasma oxidation rates
to be elevated in AD. These data implicate transition
and antioxidant levels underlines the value of the in vitro
metal ions as potential oxidants for CSF lipoproteins in
measurement of oxidation as a marker for oxidative
stress in vivo. The multiple regression analysis showed
Reduction of catalytically active protein-bound tran-
that the increased oxidation levels were specifically cor-
sition metal ions must take place to initiate oxidation,
related with the presence of the disease rather than with
and reductants, such as ascorbate or tocopherol, are able
other factors. A comparison of CSF oxidation param-
to mediate reduction [34,53]. However, it has been
eters between AD patients and patients with other
shown that this particular pro-oxidative activity of ascor-
degenerative neurological diseases, such as Parkin-
bate does not results in its total pro-oxidative action on
son’s disease and amyotrophic lateral sclerosis, is
lipoprotein oxidation; the well-known radical-scaveng-
ing action of ascorbate appears to prevail and ascorbate
In conclusion, it should be noted that AD, a multifac-
remains an antioxidant even in the presence of redox-
torial disease, is neither genetically fully elucidated, nor
active transition metal ions [54,55]. The lower levels of
are all the factors influencing its pathogenesis known.
ascorbate we measured in the CSF of AD patients are
The present study indicates that increased lipoprotein
therefore in accordance with higher CSF oxidizability.
oxidation in CSF may be an additional important factor
The high oxygen consumption in the central nervous
in the progression of the disease. The recently published
system implies a potentially increased production of ox-
trial on the antioxidative treatment of patients with AD,
ygen radicals and there is a elevated requirement for
proposing a positive effect on the course of the disease,
antioxidative molecules. However, ascorbate alone is
supports this hypothesis [18]. In vitro measurements of
three to five times higher in CSF, while all lipophilic
lipoprotein oxidation in CSF might be useful as an ad-
molecules are around 100 –500 times less concentrated in
ditional tool for the clinical diagnosis of AD. They can
CSF as compared to plasma. Therefore, ascorbate might
also be of particular interest as biochemical markers for
be of special relevance in antioxidative protection of
the evaluation of antioxidant treatment of AD patients.
lipoproteins in CSF. Antioxidant levels could be ex-pected to be decreased in the CSF of AD patients as aconsequence of high levels of oxidative stress in vivo. Acknowledgements — We thank Dr. David Evans for the critical read-
Our finding that ascorbate is significantly decreased and
ing of the manuscript and Diana Daher for accurate measurement of
lipophilic antioxidants are slightly decreased among AD
some of the samples. This study was performed in the framework of theResearch Group “Molecular Pathomechanisms in Alzheimer’s Dis-
patients therefore strongly suggests that oxidation is a
ease,” which was initiated by Prof. Roger Nitsch and is supported by
current feature of AD pathology in vivo. This finding is
the Grant Ni 486/2-1 of the Deutsche Forschungsgemeinschaft. REFERENCES
lation and quantification of soluble Alzheimer’s beta-peptide from
[1] Haass, C. Presenilin because of presenilin: the presenilin genes
biological fluids. Nature 359:325–327; 1992.
and early onset Alzheimer’s disease. Curr. Opin. Neurol. 9:254 –
[22] Koudinov, A. R.; Koudinova, N. V.; Kumar, A.; Beavis, R. C.;
Ghiso, J. Biochemical characterization of Alzheimer’s soluble
[2] Hardy, J. Amyloid, the presenilins and Alzheimer’s disease.
amyloid beta protein in human cerebrospinal fluid: association
Trends. Neurosci. 20:154 –159; 1997.
with high density lipoproteins. Biochem. Biophys. Res. Commun.
[3] Smith, C. D.; Carney, J. M.; Starke Reed, P. E.; Oliver, C. N.;
223:592–597; 1996.
Stadtman, E. R.; Floyd, R. A.; Markesbery, W. R. Excess brain
[23] Koudinov, A.; Matsubara, E.; Frangione, B.; Ghiso, J. The soluble
protein oxidation and enzyme dysfunction in normal aging and in
form of Alzheimer’s amyloid beta protein is complexed to high
Alzheimer disease. Proc. Natl. Acad. Sci. USA 88:10540 –10543;
density lipoprotein 3 and very high density lipoprotein in normal
human plasma. Biochem. Biophys. Res. Commun. 205:1164 –
[4] Dyrks, T.; Dyrks, E.; Hartmann, T.; Masters, C.; Beyreuther, K.
Amyloidogenicity of beta A4 and beta A4-bearing amyloid pro-
[24] Pitas, R. E.; Boyles, J. K.; Lee, S. H.; Hui, D.; Weisgraber, K. H.
tein precursor fragments by metal-catalyzed oxidation. J. Biol.
Lipoproteins and their receptors in the central nervous system. Chem. 267:18210 –18217; 1992. J. Biol. Chem. 262:14352–14360; 1987.
[5] Mark, R. J.; Blanc, E. M.; Mattson, M. P. Amyloid beta-peptide
[25] Montine, K. S.; Bassett, C. N.; Ou, J. J.; Markesbery, W. R.;
and oxidative cellular injury in Alzheimer’s disease. Mol. Neuro-
Swift, L. L.; Montine, T. J. Apolipoprotein E allelic influence on
biol. 12:211–224; 1996.
human cerebrospinal fluid apolipoproteins. J. Lipid Res. 39:2443–
[6] Markesbery, W. R. Oxidative stress hypothesis in Alzheimer’s
disease. Free Radic. Biol. Med. 23:134 –147; 1997.
[26] Steinberg, D.; Parthasarathy, S.; Carew, T. E.; Khoo, J. C.;
[7] Multhaup, G.; Ruppert, T.; Schlicksupp, A.; Hesse, L.; Beher, D.;
Witztum, J. L. Beyond cholesterol. N. Engl. J. Med. 320:915–
Masters, C. L.; Beyreuther, K. Reactive oxygen species and
Alzheimer’s disease. Biochem. Pharmacol. 54:533–539; 1997.
[27] Montine, T. J.; Montine, K. S.; Swift, L. L. Central nervous
[8] Spector, R. Vitamin homeostasis in the central nervous system.
system lipoproteins in Alzheimer’s disease. Am. J. Pathol. 151: N. Engl. J. Med. 296:1393–1398; 1977.
[9] Vatassery, G. T.; Nelson, M. J.; Maletta, G. J.; Kuskowski, M. A.
[28] Arlt, S.; Finckh, B.; Beisiegel, U.; Kontush, A. Time-course of
Vitamin E (tocopherols) in human cerebrospinal fluid. Am. J.
oxidation of lipids of human cerebrospinal fluid. Free Radic. Res.Clin. Nutr. 53:95–99; 1991.
[10] Smith, M. A.; Richey, P. L.; Taneda, S.; Kutty, R. K.; Sayre,
[29] McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price,
L. M.; Monnier, V. M.; Perry, G. Advanced Maillard reaction end
D.; Stadlan, E. M. Clinical diagnosis of Alzheimer’s disease:
products, free radicals, and protein oxidation in Alzheimer’s
report of the NINCDS-ADRDA Work Group under the auspices
disease. Ann. N.Y. Acad. Sci. 738:447– 454; 1994.
of Department of Health and Human Services Task Force on
[11] Smith, M. A.; Sayre, L. M.; Vitek, M. P.; Monnier, V. M.; Perry,
Alzheimer’s Disease. Neurology 34:939 –944; 1984.
G. Early ageing and Alzheimer’s. Nature 374:316; 1995.
[30] Esterbauer, H.; Striegl, G.; Puhl, H.; Rotheneder, M. Continuous
[12] Montine, K. S.; Olson, S. J.; Amarnath, V.; Whetsell, W. O., Jr.;
monitoring of in vitro oxidation of human low density lipoprotein.
Graham, D. G.; Montine, T. J. Immunohistochemical detection of
Free Radic. Res. Commun. 6:67–75; 1989.
4-hydroxy-2-nonenal adducts in Alzheimer’s disease is associated
[31] Lenz, M. L.; Hughes, H.; Mitchell, J. R.; Via, D. P.; Guyton, J. R.;
with inheritance of APOE4. Am. J .Pathol. 150:437– 443; 1997.
Taylor, A. A.; Gotto, A. M. J.; Smith, C. V. Lipid hydroperoxy
[13] Sayre, L. M.; Zelasko, D. A.; Harris, P. L.; Perry, G.; Salomon,
and hydroxy derivatives in copper-catalyzed oxidation of low
R. G.; Smith, M. A. 4-Hydroxynonenal-derived advanced lipid
density lipoprotein. J. Lipid Res. 31:1043–1050; 1990.
peroxidation end products are increased in Alzheimer’s disease.
[32] Regnstrom, J.; Strom, K.; Moldeus, P.; Nilsson, J. Analysis of
J. Neurochem. 68:2092–2097; 1997.
lipoprotein diene formation in human serum exposed to copper.
[14] Lovell, M. A.; Ehmann, W. D.; Butler, S. M.; Markesbery, W. R. Free Radic. Res. Commun. 19:267–278; 1993.
Elevated thiobarbituric acid-reactive substances and antioxidant
[33] Kontush, A.; Beisiegel, U. Measurement of oxidizability of blood
enzyme activity in the brain in Alzheimer’s disease. Neurology
plasma. Methods Enzymol. 299:35– 49; 1999. 45:1594 –1601; 1995.
[34] Kontush, A.; Meyer, S.; Finckh, B.; Kohlschutter, A.; Beisiegel,
[15] Hershey, C. O.; Hershey, L. A.; Varnes, A.; Vibhakar, S. D.;
U. Alpha-tocopherol as a reductant for Cu(II) in human lipopro-
Lavin, P.; Strain, W. H. Cerebrospinal fluid trace element content
teins. J. Biol. Chem. 271:11106 –11112; 1996.
in dementia: clinical, radiologic, and pathologic correlations.
[35] Finckh, B.; Kontush, A.; Commentz, J.; Hubner, C.; Burdelski,
Neurology 33:1350 –1353; 1983.
M.; Kohlschutter, A. Sensitive HPLC techniques for the simulta-
[16] Dedman, D. J.; Treffry, A.; Candy, J. M.; Taylor, G. A.; Morris,
neous determination of tocopherols, tocotrienols, ubiquinols and
C. M.; Bloxham, C. A.; Perry, R. H.; Edwardson, J. A.; Harrison,
ubiquinones in biological samples. Methods Enzymol. 299:341–
P. M. Iron and aluminium in relation to brain ferritin in normal
individuals and Alzheimer’s-disease and chronic renal-dialysis
[36] Barja, G.; Hernanz, A. Vitamin C, dehydroascorbate, and uric
patients. Biochem. J. 287:509 –514; 1992.
acid in tissues and serum: high-performance liquid chromatogra-
[17] Multhaup, G.; Schlicksupp, A.; Hesse, L.; Beher, D.; Ruppert, T.;
phy. Methods Enzymol. 234:331–337; 1994.
Masters, C. L.; Beyreuther, K. The amyloid precursor protein of
[37] Spranger, T.; Finckh, B.; Fingerhut, R.; Kohlschutter, A.; Beisie-
Alzheimer’s disease in the reduction of copper(II) to copper(I).
gel, U.; Kontush, A. How different constituents of human plasma
Science 271:1406 –1409; 1996.
and low density lipoprotein determine plasma oxidizability by
[18] Sano, M.; Ernesto, C.; Thomas, R. G.; Klauber, M. R.; Schafer,
copper. Chem. Phys. Lipids 91:39 –52; 1998.
K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C. W.;
[38] Hixson, J. E.; Vernier, D. T. Restriction isotyping of human
Pfeiffer, E.; Schneider, L. S.; Thal, L. J. A controlled trial of
apolipoprotein E by gene amplification and cleavage with HhaI. J.
selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s
Lipid Res. 31:545–548; 1990.
disease N. Engl. J. Med. 336:1216 –1222; 1997.
[39] Roses, A. D.; Saunders, A. M. APOE is a major susceptibility
[19] Behl, C.; Davis, J.; Cole, G. M.; Schubert, D. Vitamin E protects
gene for Alzheimer’s disease. Curr. Opin. Biotechnol. 5:663–
nerve cells from amyloid beta protein toxicity. Biochem. Biophys.Res. Commun. 186:944 –950; 1992.
[40] Lonnrot, K.; Metsa Ketela, T.; Molnar, G.; Ahonen, J. P.; Latvala,
[20] Davis, J. B. Oxidative mechanisms in beta-amyloid cytotoxicity.
M.; Peltola, J.; Pietila, T.; Alho, H. The effect of ascorbate and
Neurodegeneration. 5:441– 444; 1996.
ubiquinone supplementation on plasma and CSF total antioxidant
[21] Seubert, P.; Vigo, P. C.; Esch, F.; Lee, M.; Dovey, H.; Davis, D.;
capacity. Free Radic. Biol. Med. 21:211–217; 1996.
Sinha, S.; Schlossmacher, M.; Whaley, J.; Swindlehurst, C. Iso-
[41] Heinecke, J. W. Mechanisms of oxidative damage of low density
lipoprotein in human atherosclerosis. Curr. Opin. Lipidol. 8:268 –
[52] Sayre, L. M.; Perry, G.; Smith, M. A. Redox metals and neuro-
degenerative disease. Curr. Opin. Chem. Biol. 3:220 –225; 1999.
[42] Borghini, I.; Barja, F.; Pometta, D.; James, R. W. Characterization
[53] Lynch, S. M.; Frei, B. Reduction of copper, but not iron, by
of subpopulations of lipoprotein particles isolated from human
human low density lipoprotein (LDL). J. Biol. Chem. 270:5158 –
cerebrospinal fluid. Biochim. Biophys. Acta 1255:192–200; 1995.
[43] Frei, B.; Gaziano, J. M. Content of antioxidants, preformed lipid
[54] Berger, T. M.; Polidori, M. C.; Dabbagh, A.; Evans, P. J.; Halli-
hydroperoxides, and cholesterol as predictors of the susceptibility
well, B.; Morrow, J. D.; Roberts, L. J.; Frei, B. Antioxidant
of human LDL to metal ion-dependent and -independent oxida-
activity of vitamin C in iron-overloaded human plasma. J. Biol.
tion. J. Lipid Res. 34:2135–2145; 1993. Chem. 272:15656 –15660; 1997.
[44] Kontush, A.; Hubner, C.; Finckh, B.; Kohlschutter, A.; Beisiegel,
[55] Kontush, A.; Finckh, B.; Karten, B.; Kohlschutter, A.; Beisiegel,
U. How different constituents of low density lipoprotein deter-
U. Antioxidant and prooxidant activity of alpha-tocopherol in
mine its oxidizability by copper: a correlational approach. Free
human plasma and low density lipoprotein. J. Lipid Res. 37: Radic. Res. 24:135–147; 1996.
[45] Loeffler, D. A.; DeMaggio, A. J.; Juneau, P. L.; Brickman, C. M.;
[56] Subbarao, K. V.; Richardson, J. S.; Ang, L. C. Autopsy samples
Mashour, G. A.; Finkelman, J. H.; Pomara, N.; LeWitt, P. A.
of Alzheimer’s cortex show increased peroxidation in vitro.
Ceruloplasmin is increased in cerebrospinal fluid in Alzheimer’s
J. Neurochem. 55:342–345; 1990.
disease but not Parkinson’s disease. Alzheimer. Dis. Assoc. Dis- ord. 8:190 –197; 1994.
[46] Lamb, D. J.; Leake, D. S. Iron released from transferrin at acidic
ABBREVIATIONS
pH can catalyse the oxidation of low density lipoprotein. FEBS Lett. 352:15–18; 1994.
[47] Mukhopadhyay, C. K.; Ehrenwald, E.; Fox, P. L. Ceruloplasmin
enhances smooth muscle cell- and endothelial cell-mediated lowdensity lipoprotein oxidation by a superoxide-dependent mecha-
nism. J. Biol. Chem. 271:14773–14778; 1996.
AAPH—2,2Ј-azobis-(2-amidinopropane) hydrochloride
[48] Kennard, M. L.; Feldman, H.; Yamada, T.; Jefferies, W. A. Serum
levels of the iron binding protein p97 are elevated in Alzheimer’s disease. Nat. Med. 2:1230 –1235; 1996.
[49] Loeffler, D. A.; Connor, J. R.; Juneau, P. L.; Snyder, B. S.;
Kanaley, L.; DeMaggio, A. J.; Nguyen, H.; Brickman, C. M.;
LeWitt, P. A. Transferrin and iron in normal, Alzheimer’s disease, and Parkinson’s disease brain regions. J. Neurochem. 65:710 –
HPLC— high-performance liquid chromatography
[50] Loeffler, D. A.; LeWitt, P. A.; Juneau, P. L.; Sima, A. A.;
Nguyen, H. U.; DeMaggio, A. J.; Brickman, C. M.; Brewer, G. J.;Dick, R. D.; Troyer, M. D.; Kanaley, L. Increased regional brain
concentrations of ceruloplasmin in neurodegenerative disorders. Brain Res. 738:265–274; 1996.
[51] Smith, M. A.; Harris, P. L.; Sayre, L. M.; Perry, G. Iron accu-
mulation in Alzheimer disease is a source of redox-generated free
radicals. Proc. Natl. Acad. Sci. USA 94:9866 –9868; 1997.
2000-2002 Contributions 2002 – Present Dean Kereiakes. Editorial Board, Circulation Dean Kereiakes. Editorial Board – Reviews in Cardiovascular Dean Kereiakes. Editorial Board, American Journal of Cardiology Bin JP, Pelberg RA , Wei K, Le DE, Goodman NC, Kaul S. Dobutamine versus dipyridamole for inducing reversible perfusion defects in chronic multivessel coronary artery stenosi
Atomoxetina (ATX) La atomoxetina (ATX) (Strattera®) es el único fármaco no-estimulante aprobado para el tratamiento de TDAH. Mecanismo de acción La ATX bloquea el transportador de la noradrenalina (NET) presináptico, de manera selectiva y potente, impidiendo la recaptación de NA a la neurona presináptica, y por tanto aumentando la concentración de NA en todo el cerebro. Adem�

 Free Radical Biology & Medicine, Vol. 28, No. 3, pp. 351–360, 2000
Copyright 2000 Elsevier Science Inc.
Free Radical Biology & Medicine, Vol. 28, No. 3, pp. 351–360, 2000
Copyright 2000 Elsevier Science Inc.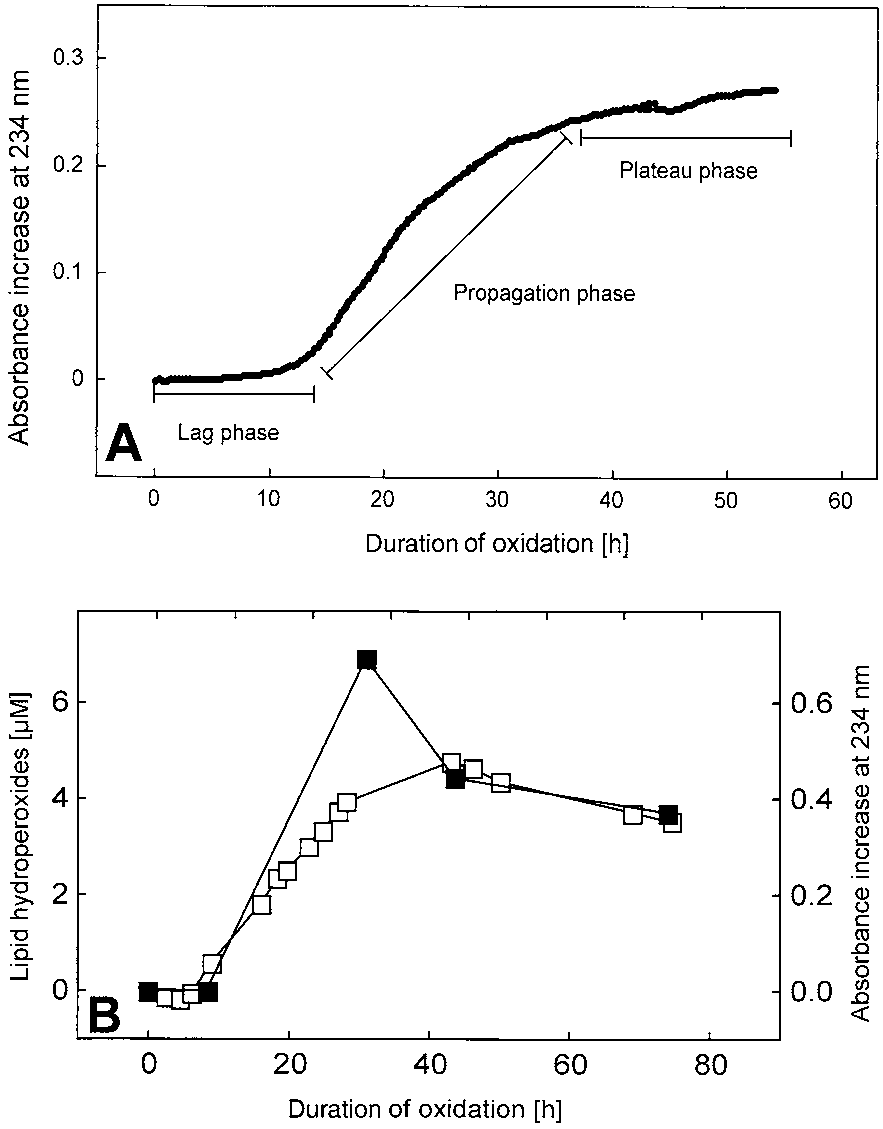
 * p Ͻ .01, † p Ͻ .05 vs. corresponding control group.
* p Ͻ .01, † p Ͻ .05 vs. corresponding control group.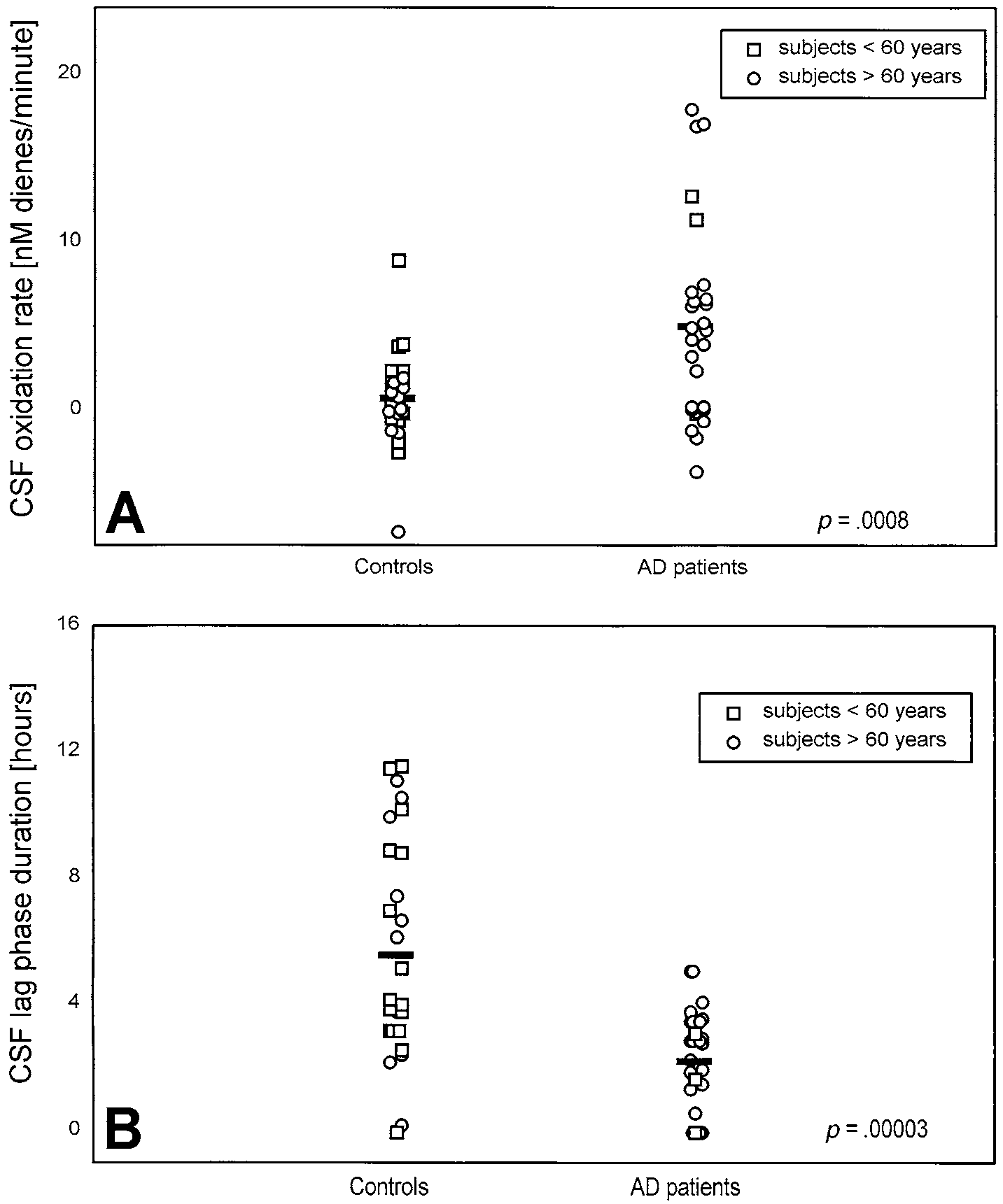 Fig. 3. (A) Rate and (B) lag phase of autoxidation of CSF obtained from AD patients and control subjects. CSF was diluted 10-foldwith PBS and incubated at 37°C in the absence of exogenous oxidants. Bars correspond to the mean values calculated for each group.
Fig. 3. (A) Rate and (B) lag phase of autoxidation of CSF obtained from AD patients and control subjects. CSF was diluted 10-foldwith PBS and incubated at 37°C in the absence of exogenous oxidants. Bars correspond to the mean values calculated for each group.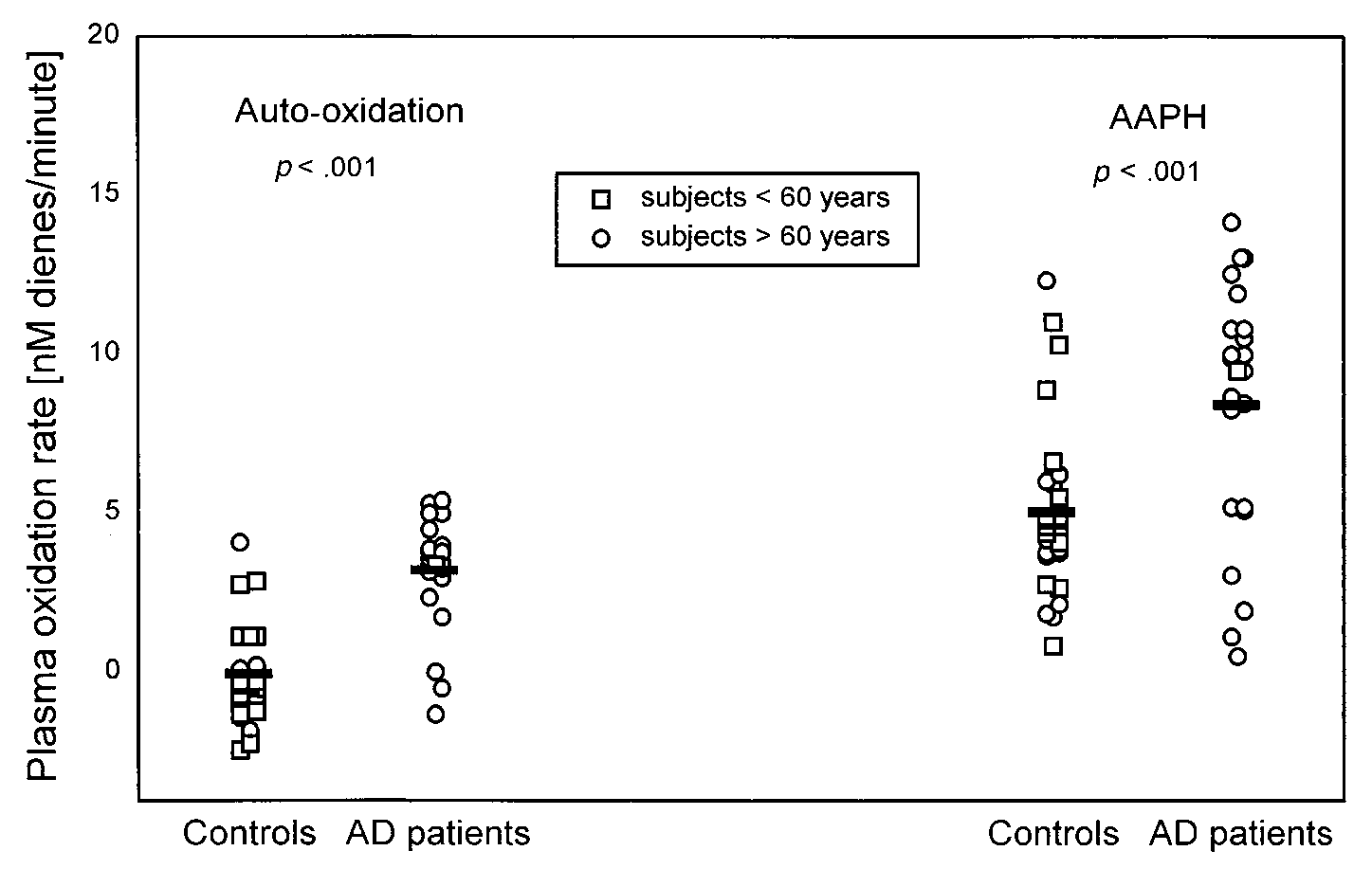 Table 2. Protein, Lipids, and Antioxidants in CSF of AD Patients and Control Subjects
a Weight percentage of TFA. * p Ͻ .001; † p Ͻ .01; ‡ p Ͻ .05 vs. corresponding control group.
Table 2. Protein, Lipids, and Antioxidants in CSF of AD Patients and Control Subjects
a Weight percentage of TFA. * p Ͻ .001; † p Ͻ .01; ‡ p Ͻ .05 vs. corresponding control group.