Tadalafil appartiene alla classe degli inibitori selettivi della fosfodiesterasi di tipo 5, con un profilo farmacocinetico caratterizzato da un’emivita terminale di circa diciotto ore. Dopo somministrazione orale viene assorbito rapidamente e raggiunge concentrazioni plasmatiche massime in due ore. La biotrasformazione avviene principalmente tramite CYP3A4 con formazione di metaboliti inattivi, escreti in prevalenza con le feci. L’elevato legame alle proteine plasmatiche (>90%) assicura una distribuzione stabile. Nei confronti delle altre molecole della stessa classe, cialis compresse italia è noto per la durata prolungata dell’attività farmacologica.
Pii: s0166-1280(03)00307-5
Journal of Molecular Structure (Theochem) 632 (2003) 297–307
How important is the refinement of transition state structures
` ngels Gonza´lez-Lafont, Jose´ M. Lluch*
Departament de Quı´mica, Universitat Auto`noma de Barcelona, Bellaterra, Barcelona 08193, Spain
Received 30 October 2002; revised 18 December 2002; accepted 18 December 2002
In this paper the need to use a second derivatives direct algorithm to refine the location of transition state structures obtained
in enzymatic systems has been analyzed. The 25 approximate QM/MM transition state structures previously found by means ofa reaction coordinate approach for the three mechanisms of racemization of mandelate and propargylglycolate by mandelateracemase enzyme have been refined using a modified micro-iterative optimization method developed in this work. Therefinement of transition state structures is especially useful to assure that a structure, found as the highest potential energy pointon a profile depicted by a particular reaction coordinate, lies in the correct quadratic region. This is more important in thosesteps of the enzymatic process where the selected reaction coordinate may not reflect quite accurately the geometrical changestaking place in the active site.
q 2003 Elsevier B.V. All rights reserved.
Keywords: Quantum mechanical/molecular mechanical transition state structures; Reaction coordinate approach; Micro-iterative method;Second derivatives direct optimization method; Mandelate racemase reaction mechanisms
the enzyme – substrate complex for a fixed positionof the nuclei. That is, using this technique a small
Enzymes speed up reactions by many orders of
region at the active site of an enzyme is described
magnitude using fundamental physical processes to
quantum mechanically, whereas the surrounding
increase chemical reactivity. Methods that permit to
protein is included by a simpler MM representation.
calculate and to explore the Potential Energy
Classical molecular dynamics simulations have to be
Surface (PES) of an enzymatic reaction are needed
carried out to sample extensively the configurationspace, looking for new regions of the PES around
to understand theoretically the enormous catalytic
minimum energy structures representing possible
power of enzymes. A hybrid quantum mechanical/
reactant and product complexes. However, both
energetic and entropic factors make it impossible in
can be used to obtain the potential energy of
practice the molecular dynamics generation ofreactive trajectories going from the reactant region
* Corresponding author. Tel.: þ34-93-581-2138; fax: þ34-93-
to the product region in a canonical ensemble at a
E-mail address: [email protected] (J.M. Lluch).
given temperature. Then kinetics information can be
0166-1280/03/$ - see front matter q 2003 Elsevier B.V. All rights reserved. doi:10.1016/S0166-1280(03)00307-5
X. Prat-Resina et al. / Journal of Molecular Structure (Theochem) 632 (2003) 297–307
obtained by means of the Transition State Theory, a
PES to locate the set of stationary points that
statistical approach to real dynamics, which is able
connects with the reactant and the product by
to provide canonical rate constants kðTÞ .
means of the real reaction pathway should be
According to Variational Transition State Theory
highly recommended prior to the free energy
the canonical rate constant depends on the
calculations. So, the set of dividing surfaces should
generalized free energy barrier, that is, the maxi-
be raised along that real reaction pathway.
mum value of the generalized free energies
The point now is how precise the location of the
associated with a set of dividing surfaces built up
transition state structures has to be to produce a
along a suitable reaction pathway taken as a
reliable reaction path. Is the location of the transition
reference. The generalized free energies can be
state structure as the maximum energy point along an
obtained, for instance, from molecular dynamics
energy profile built up as a function of a conveniently
simulations using the umbrella sampling technique
chosen reaction coordinate sufficient or after that the
with an adequate biasing potential or by means of
transition state structures have to be refined? The
statistical perturbation theory Once the reactant
purpose of this paper is to use the different reaction
and the product have been localized, a progress
channels we have previously found for the
coordinate connecting them and based on suitable
racemization of mandelate and propargylglycolate by
internal coordinates can be adopted to define the
mandelate racemase enzyme to shed some light to that
reaction pathway. As an alternative, knowing the
valence bond structures of both reactant and
This paper is structured as follows. In Section 2 we
product, a mapping potential as a function of
briefly review the methods used for locating transition
the diagonal elements of an empirical valence
state structures in enzymatic catalysis, in Section 3 we
present the enzymatic system where we have tested
define a reaction pathway as a collective reaction
the different methods, and in Section 4 we compare
coordinate analogous to the solvent coordinate used
the results obtained with the different strategies. A
in Marcus theory for electron transfer reactions. The
EVB provides a very effective way of exploring thePES of a substrate – enzyme complex between anyset of possible intermediates. Thus it is essential todefine the problem in terms of feasible reactants,
2. Methods to locate stationary points in big
products and intermediates. In all cases, nuclear
quantum effects and corrections accounting for therecrossing of the dividing surface can be introduced
Using QM/MM potentials the energy calculation
is expensive enough to look for effective methods
that reach convergence with as minimum number
approaches become very fruitful, their practical
of steps as possible. On the other hand, the high
implementation is generally based on a reference
number of degrees of freedom in enzymatic
path constructed using the information extracted
systems makes the usage of standard second order
from the reactant and product. However, this
methods, such as Newton – Raphson, too compu-
procedure could lead to inaccurate results. Due to
tational demanding due to the construction and
the complexity of the enzymatic reactions, the real
manipulation of a very big second derivatives
reaction pathway can be very different from the
matrix (Hessian). Different strategies have already
apparent one at first glance. The enzymatic reaction
been applied to locate transition state structures in
can actually take place through several parallel and
such big systems All of them try to find a
kinetically competitive channels, each one consist-
compromise between effectiveness and low compu-
ing of multiple steps, involving several intermedi-
tational cost. On the other hand, although the usage
ates in going from the reactant to the product, then
of suitable internal coordinates is an alternative
leading to a priori unexpected reaction paths. As a
way to achieve converged structures effectively in
consequence, an exploration of the corresponding
X. Prat-Resina et al. / Journal of Molecular Structure (Theochem) 632 (2003) 297–307
optimization with Cartesian coordinates will beconsidered.
The easiest procedure to look for transition state
structures is to freeze one degree of freedom of oursystem, a coordinate (e.g. a distance, angle or dihedralor a combination of them) that is representative of thereaction we are studying, and to perform restrainedminimizations scanning this coordinate from reactantsvalue to products all along what it is supposed to bethe reaction path. In this case we obtain a discreteenergy profile and the highest point of this profile istaken as the transition state structure of the reaction. The crucial aspect is to choose an adequate reactioncoordinate and to perform the scan with as manypoints as possible. It is not always so intuitive tochoose such a coordinate, and even when thecoordinate is the right one we will show that insome cases the proposed transition state is an incorrectstructure.
In our test cases we have used an harmonic
potential with a constant of 10,000 kcal/mol/A
restrain the chosen coordinate during the minimiz-ation that is performed with the LBFGS method
Fig. 1. Scheme for the location of transition state structures in
In this paper the distance between the acceptor atom
enzymatic systems moving only a core while keeping the rest of
and the hydrogen that is being transferred in each step
environment atoms frozen (top). Micro-iterative scheme (bottom).
has been always used to define the reactioncoordinate.
A next step is the usage of methods that knowing
small part of the system (core) is participating in
the reactant and product structure make use of the
the reaction, then keeping the rest of the degrees of
energy and its first derivatives to find the transition
state structure and an approximation to the minimum
NR or RFO search is performed only for this core,
energy path To our knowledge only Conjugate
avoiding the environment to relax. The intuitive
Peak Refinement method has been applied to
next solution is permitting the environment to relax
using an inexpensive optimization method (e.g. Conjugate Gradient LBFGS ABNR
while in the core a transition state structure searchis performed with the NR-like method. Both
Another strategy consists in the direct location
processes are carried out alternating one and the
of the transition state structure with methods that
other until self-consistency (This is the
use the second derivatives of the energy (e.g.
and is the method that we will consider in our
comparative study as the approach that gives the
tational cost mentioned above the treatment of the
most refined structure. This so-called micro-itera-
whole set of degrees of freedom at the same level
tive method can be achieved in several manners
is not feasible. In a first approximation only a
depending on the minimizer for the environment,
X. Prat-Resina et al. / Journal of Molecular Structure (Theochem) 632 (2003) 297–307
the interaction between core and environment, andhow often the environment must be relaxed duringthe transition state structure search in the core. Allthese features will not be discussed here.
The method that we have implemented makes use
of the RFO technique for the location of the transition
Fig. 2. Racemization reaction of mandelate.
state structure in the core region The minimiz-
norm of the whole system is converged with a
ation of the environment is carried out with LBFGS
and it is performed every time the RFO convergesinstead of at every step of this transition state structure
The reaction coordinate method and the micro-
search. The interaction between the core and the
iterative procedure have been implemented in the
environment during the minimization of the latter is
always the full QM/MM energy at every step. That is,
we do not calculate ESP charges and fix it all along the
the transition state structures of the reaction mechan-
minimization process like other authors have done
isms of the enzymatic system that is presented in
In our case this strategy was easier to implement,
permits to obtain a real interaction between both
The common procedure has been to take the
regions and gives the possibility of including some
structures obtained with the reaction coordinate
quantum mechanical atoms in the environment zone.
method as the input for the micro-iterative method.
This method can easily find a transition state
structure with low computational cost. If the initialstructure is appropriate it is faster than the reaction
coordinate method mentioned above. When the coreis not selected adequately and some atoms of the
3.1. The reaction, the different substrates
environment zone participate directly or indirectly in
the reaction, then some coupling between the twozones might appear that avoids to reach the desired
Mandelate racemase catalyzes the reversible iso-
convergence. The solution to this problem is the
merization of both enantiomers of mandelate
selection of a bigger core zone. A bigger core zone
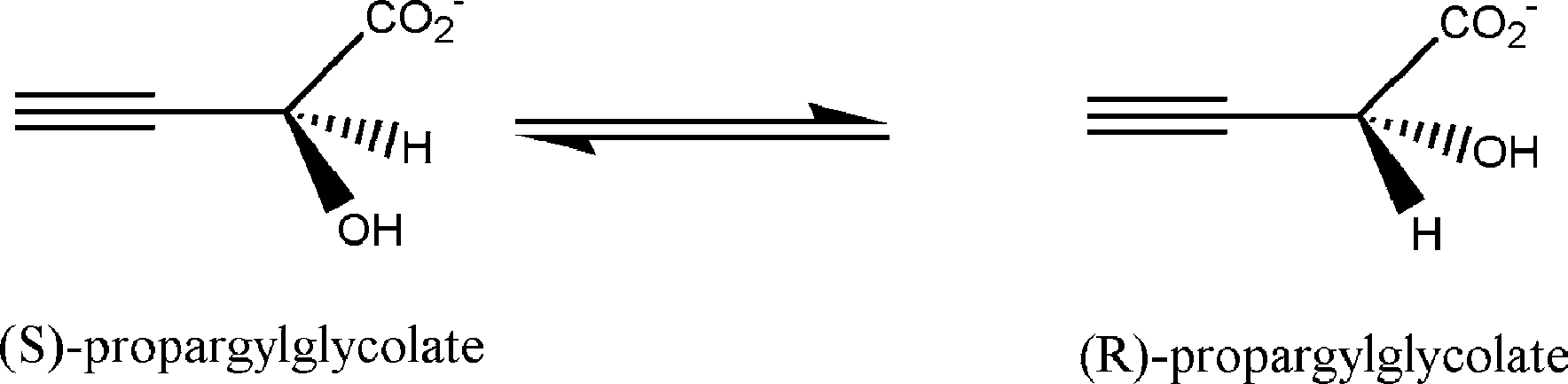
In addition, propargylglycolate () has been
implies the calculation and manipulation of a bigger
found to be a moderately good substrate for
Hessian matrix. The drawbacks can be solved with
racemization. Both substrates can evolve from
an approximated initial Hessian matrix and a partial
reactants to products through three parallel mechan-
diagonalization instead of a full diagonalization
isms . The residues that belong to the active center
(note that RFO only needs one eigenvector to
calculate the displacement; this partial diagonaliza-
tion can be carried out with the standard LAPACK
change that occurs at each step of the mechanisms. We
will consider the enantiomer (S) as the reactant and the
The micro-iterative scheme we have used in this
(R) as the product. For propargylglycolate both
mechanisms I and II require a previous proton transfer,through the transition state (TS) structure 1, from the
1. Minimization of the selected environment with
auxiliary proton donors Lys164 (mechanism I) or
LBFGS keeping the core frozen until convergence.
2. Transition state structure search with RFO moving
only the atoms of the core. This process is carriedout until convergence as well.
3. Check for the gradient norm of the total system:
if it is not minimized, it returns to point 1. Theprocess will not be finished until the gradient
Fig. 3. Racemization reaction of propargylglycolate.
X. Prat-Resina et al. / Journal of Molecular Structure (Theochem) 632 (2003) 297–307
transfer neither from Lys164 nor Glu317 beingrequired.
For mandelate mechanism I consists of the same
six steps with the same transition state structures as inmechanism I for propargylglycolate. Mechanism IIconsist of only five steps because the change ofconfiguration of the Ca atom of the substrate takesplace in a concerted way with the proton donationfrom His297 at TS5 (TS4 does not exist in this case). Mechanism III for mandelate involves formally twosteps, but the well between the two correspondingtransition state structures is so shallow that, for the
Fig. 4. Mandelate racemase active center. The X symbol stands for
purpose of this paper, it can be considered as a one-
the phenyl or ethinyl groups when the substrates are mandelate or
Glu317 (mechanism II). In both mechanisms the
It has to be underlined that the description of the
proton attached to the Ca atom (the one adjacent to
different steps is extremely oversimplified. In chemi-
the carboxylate group) of the substrate is being
cal systems so complicated like enzymes, many atoms
abstracted by Lys166 at TS2. His297 is migrating at
belonging to a lot of residues move in a significant
TS3, approaching to the substrate and moving away
way in each step, accompanying the main change that
from Glu247. The Ca atom of the substrate, the
defines that step. For instance, many atoms have to
stereogenic center, is changing its configuration from
readapt smoothly their positions to the new charge
(S) to (R), through a sp2 hybridisation, at TS4. A proton
distribution along a proton tranfer, or, conversely,
transfer from His297 to the Ca atom of the substrate is
some residues have to migrate prior to a proton
occurring at TS5. Finally, the proton transfer that
happened in the first step is reversed through TS6. Incontrast, mechanism III for propargylglycolate is an
3.2. The theoretical model used: the potential energy
asynchronic concerted mechanism which involves
proton abstraction by Lys166, configuration changeof the Ca atom of the substrate and protonation from
For the sake of comparison with the results
His297 within a unique step, no previous proton
already published, the enzymatic model used here
Fig. 5. Mandelate racemase mechanisms (see text). Mechanisms I and II need the previous protonation of the carboxyl group of the substratebefore the isomerization. Mechanism III reaches products in one concerted step. Bold type atoms belong to the substrate.
X. Prat-Resina et al. / Journal of Molecular Structure (Theochem) 632 (2003) 297–307
is the same that we used in our previous studies of
Potential energies (kcal/mol) for the racemization of mandelate
vinylglycolate substrates A more detailed
description of the method can be found there.
Here we will only mention a few aspects. Thestructure with the Protein Data Bank code 1MNS
has been used to build up our model. The zone
chosen to be represented quantum mechanically
with a semiempirical Hamiltonian (PM3 in all
constituted by the substrate, the magnesium atom, a
water coordinated to it, and the lateral chains of the
residues that participate actively in the isomeriza-
The corresponding values obtained from the micro-iterative
tion reaction (). This means 80 or 88 atoms,
hydrogen link atoms to cap the seven QM/MM
practically removed by performing a scanning with
frontiers included, for propargylglycolate or
more intermediate points along the reaction coordi-
mandelate complexes, respectively. The rest of theenzyme and solvation waters are treated with the
nate. On the contrary, in the case of propargylgly-
colate important differences exist among some
that a total of 3963 atoms will constitute the whole
energy barriers obtained with the two methods.
These differences are mainly centered in step 4,
The region corresponding to the moving atoms
when stereogenic center Ca changes its configur-
includes all the residues that fall into a sphere of 15 A
ation. Another significant discrepancy can be seen
centered at the magnesium atom. This implies 1299
at the TS1 of the mechanism I. A geometry
moving atoms for the mandelate case. In what refers
analysis of the corresponding transition state
to the micro-iterative scheme the core zone has been
structures will shed some light to this point.
chosen to include the important atoms at every step. The full QM/MM interaction between core and
environment used in this paper enables us to excludefrom the core region the QM atoms which are not
The more relevant distances for the transition state
relevant in a particular mechanism step. This fact
structures corresponding to the racemization of
permits us to work with a smaller Hessian for the core.
mandelate and propargylglycolate according to mech-
In any case, the selection of the two regions has to be
anisms I, II and III (located using the reaction
Table 2Potential energies (kcal/mol) for the racemization of propargylgly-
4. Comparison of the results obtained with
colate using the reaction coordinate method
The potential energy barriers obtained by using
the reaction coordinate and the micro-iterative
methods for the racemization of mandelate and
respectively. No significant differences are observed
among the two sets of energy barriers in the case
The corresponding values obtained from the micro-iterative
of mandelate, no other than those that could be
X. Prat-Resina et al. / Journal of Molecular Structure (Theochem) 632 (2003) 297–307
˚ ) for the transition state structures corresponding to the racemization of mandelate and propargylglycolate according
to mechanism I using the reaction coordinate method
The same distances obtained using the micro-iterative method are given in brackets.
coordinate method and the micro-iterative method)
It can be seen that there are no significant
are presented in respectively. Since many
divergences between both sets of transition state
atoms move in each step, we have also compared the
structures in most of the steps of the mechanisms. On
positions of the main residues of the active center at
the other hand, the discrepancies in general have no
the transition state structures located using the two
important consequence on the potential energy
methods. To this aim we have calculated the root
barriers: See, for instance, that a deviation of even
mean square (RMS) of the difference between the
coordinates of the atoms at the transition state
mechanism II for mandelate, along with some RMS
structures obtained employing the reaction coordinate
method and the ones located by means of the micro-
in the active center, produces a change of only
iterative method. These RMS values are shown in
2 0.31 kcal/mol in the corresponding energy barrier
for mechanisms I, II and III, respectively.
The rest of the residues of the active center give lower
The step 4 of mechanism I and II of propargyl-
values of RMS and are not included in the tables.
glycolate is a especial case in which the transition
˚ ) for the transition state structures corresponding to the racemization of mandelate and propargylglycolate according
to mechanism II using the reaction coordinate method
The same distances obtained using the micro-iterative method are given in brackets.
X. Prat-Resina et al. / Journal of Molecular Structure (Theochem) 632 (2003) 297–307
propargylglycolate points to different directions, the
conformation of the amino group in Lys166 is
corresponding to the racemization of mandelate and propargylgly-
slightly different and the Ca atom presents a
colate according to mechanism III using the reaction coordinate
different degree of configurational change. The
Asp195 residue is depicted to realize the different
conformation of Lys166. Then, we can see in thiscase that the differences are not only in the distances
associated to the transferring hydrogens (
but in the immediate surrounding. Note that
this step 4 consists basically of the configuration
change of the Ca atom, the reaction coordinate
chosen in this case (the distance between the
transferring hydrogen and the acceptor heavy
atom) not being perhaps the most adequate.
It is important to remark that the initial structure
used for each step in the micro-iterative method has
The same distances obtained using the micro-iterative method
been the one obtained in the reaction coordinate
method, and in all cases but in the step 4 (mechanismsI and II) for propargylglycolate we have found a
state structures obtained with the two methods differ
negative eigenvalue from the beginning and a
corresponding eigenvector that describes the step. In
energy difference already seen in this step as well.
step 4 the initial structure (that is, the transition state
The differences appear in residue Lys166 and in the
structure obtained from the reaction coordinate
substrate. In we can do a visual inspection of
method) had no transition vector. This means that
what is happening. That is, the hydroxyl group of
the proposed structure according to the reaction
˚ ) for the transition state structures corresponding to the racemization of mandelate and propargylglycolate according to mechanism I
X. Prat-Resina et al. / Journal of Molecular Structure (Theochem) 632 (2003) 297–307
˚ ) for the transition state structures corresponding to the racemization of mandelate and propargylglycolate according to mechanism II
coordinate method was away from the quadratic
of the refinement of the coordinates and the potential
region with the suitable curvature corresponding to
energy of the located structure but with the problem of
the actual transition state structure of this step. So in
a transition state structure found by the reaction
this case we are not just dealing with the convenience
coordinate method that may not fulfill the adequate
˚ ) for the transition state structure corresponding to the
racemization of mandelate and propargylglycolate according tomechanism III
Fig. 6. Transition state structures located with the reaction coordinate
method (upper structure) and the micro-iterative method (lower structure)
for the step 4 in mechanism I of the substrate propargylglycolate.
X. Prat-Resina et al. / Journal of Molecular Structure (Theochem) 632 (2003) 297–307
mathematical conditions to be considered at least, as
the application of a second derivatives direct method,
an approximation to the real transition state structure
like the one presented here, is always recommended,
rather to warrant the real nature of transition state of
the located structure (in other words, that the located
propargylglycolate, the main divergence between
structure lies on the quadratic region corresponding to
both transition state structures comes from the two
the actual transition state structure), than to refine the
distances associated with the transferring hydrogen
concrete values of the potential energy barriers and
oxygen atom of the substrate: 1.451 and 1.056 A
Indeed this work concerns just to a particular
for the Lys166-N· · ·H and the H· · ·OOC distances,
enzymatic reaction. However, we have tested the
respectively, according to the reaction coordinate
racemization of two substrates, that takes place
through three different mechanisms, involving many
arising from the micro-iterative method. These
distinct steps. In all, 25 transition states have been
differences, which could be avoided using a
located, which provide a critical mass of information,
denser grid along the reaction coordinate method,
probably enough to think that the conclusions of this
leads to the corresponding potential energy barrier
paper can be quite general for the enzymatic reactions
In this paper we have built up a new second
We are grateful for financial support from the
derivatives direct method to locate transition state
Spanish ‘Ministerio de Ciencia y Tecnologı´a’ and the
structures in enzymatic reactions that differs in some
‘Fondo Europeo de Desarrollo Regional’ through
points from previous related algorithms. This method,
project No. BQU2002-00301, and the use of the
here called micro-iterative method, is based on a RFO
computational facilities of the CESCA.
procedure for the location of the transition statestructure in a previously defined core region, and onan LBFGS minimization of the environment. It has
been implemented in the module Roar within theAMBER 5.0 package. We have used this method to
[1] A. Warshel, M. Levitt, J. Mol. Biol. 103 (1976) 227.
refine the approximate QM/MM transition state
[2] M.J. Field, P.A. Bash, M. Karplus, J. Comput. Chem. 11
structures obtained using a reaction coordinate
method for the racemization of mandelate and
[3] J. Gao, M.A. Thompson (Eds.), Combined Quantum Mech-
propargylglycolate by mandelate racemase enzyme.
anical and Molecular Mechanical Methods, ACS SymposiumSeries 712, American Chemical Society, Washington, DC,
The results show that the transition state structures
located by means of the reaction coordinate method
[4] M.J. Field, J. Comput. Chem. 23 (2002) 48.
are, in general, good enough to define a reliable
[5] J. Gao, D. Truhlar, Annu. Rev. Phys. Chem. 53 (2002) 467.
reaction path of the reaction, taking into account both
[6] D. Truhlar, B. Garrett, S. Klippenstein, J. Phys. Chem. 100
the potential energy barriers and the geometrical
[7] A.R. Leach, Molecular Modelling. Principles and Appli-
structures, provided that a suitable reaction coordinate
cations, second ed., Pearson Education, England, 2001.
has been defined for each step of the reaction.
[8] A. Warshel, Computer Modeling of Chemical Reactions in
However, if the geometrical parameters chosen to
Enzymes and Solutions, Wiley, New York, 1992.
define the reaction coordinate in a concrete step do not
[9] J. Aqvist, A. Warshel, Chem. Rev. 93 (1993) 2523.
involve the main geometrical changes that take place
[10] D.G. Truhlar, J. Gao, C. Alhambra, M. Garcia-Viloca, J.
Corchado, M.L. Sa´nchez, J. Villa`, Acc. Chem. Res. 35 (2002)
in that step, the transition state structure supplied by
the reaction coordinate method can turn out to be quite
[11] C. Alhambra, J. Corchado, M.L. Sa´nchez, M. Garcia-Viloca, J.
different from the actual one. In this sense
Gao, D.G. Truhlar, J. Phys. Chem. B 105 (2001) 11326.
X. Prat-Resina et al. / Journal of Molecular Structure (Theochem) 632 (2003) 297–307
[12] X. Prat-Resina, M. Garcia-Viloca, A. Gonza´lez-Lafont, J.M.
[28] R.J. Hall, S.A. Hindle, N.A. Burton, I.H. Hillier, J. Comput.
Lluch, Phys. Chem. Chem. Phys. 4 (2002) 5365.
[13] G. Monard, X. Prat-Resina, A. Gonza´lez-Lafont, J.M. Lluch,
[29] Y. Zhang, H. Liu, W. Yang, J. Chem. Phys. 112 (2000)
Int. J. Quantum Chem. 93 (2003) 229.
[14] K. Ne´meth, O. Coulaud, G. Monard, J.G. A
Frisch, J. Comput. Chem. 24 (2003) 760.
[15] S.R. Billeter, A.J. Turner, W. Thiel, Phys. Chem. Chem. Phys.
[31] X. Prat-Resina, M. Garcia-Viloca, G. Monard, A. Gonza´lez-
Lafont, J.M. Lluch, J.M. Anglada, J.M. Bofill, Theor. Chem.
[16] B. Paizs, J. Baker, S. Suhai, P. Pulay, J. Chem. Phys. 113
[32] E. Anderson, Z. Bai, C. Bischof, S. Blackford, J. Demmel, J.
[17] D.C. Liu, J. Nocedal, Math. Programming (1989) 503.
Dongarra, J.D. Croz, A. Greenbaum, S. Hammarling,
[18] G. Henkelman, G. Jo´hannesson, H. Jo´nsson, Methods for
A. McKenney, D. Sorensen, LAPACK User’s Guide, third
finding saddle points and minimum energy path, in: S.D.
Schwartz (Ed.), Progress on Theoretical Chemistry and
[33] A. Cheng, R.S. Stanton, J.J. Vincent, A. van der Vaart, K.V.
Damodaran, S.L. Dixon, D.S. Hartsough, M. Mori, S.A. Best,
[19] S. Fischer, M. Karplus, Chem. Phys. Lett. 194 (1992) 252.
G. Monard, M. Garcia-Viloca, L.C.V. Zant, J.K.M. Merz,
[20] Q. Cui, M. Elstner, M. Karplus, J. Phys. Chem. B 106 (2002)
ROAR 2.0, The Pennsylvania State University, 1999.
[34] D. Pearlman, D. Case, J. Caldwell, W. Ross, T. Cheatham, S.
[21] Q. Cui, M. Karplus, J. Am. Chem. Soc. 124 (2002) 3093.
Debolt, D. Ferguson, G. Seibel, P. Kollman, Comput. Phys.
[22] R. Fletcher, Practical Methods of Optimization, second ed.,
[23] J. Simons, P. Jorgensen, H. Taylor, J. Ozment, J. Comput.
[35] M. Garcia-Viloca, A. Gonza´lez-Lafont, J.M. Lluch, J. Am.
[24] A. Banerjee, N. Adams, J. Simons, R. Shepard, J. Comput.
[36] M.C. Hutter, J.M. Hughes, J.R. Reimers, N.S. Hus, J. Phys.
[25] E. Besalu´, J.M. Bofill, Theor. Chem. Acc. 100 (1998) 265.
[37] N. Reuter, A. Dejaegere, B. Maigret, M. Karplus, J. Phys.
[26] B.R. Brooks, R.E. Bruccoleri, B.D. Olafson, D.J. States, S.
Swaminathan, M. Karplus, J. Comput. Chem. 4 (1983) 187.
[38] S.J. Weiner, P.A. Kollman, D.A. Case, U.C. Singh, C. Ghio,
[27] A.J. Turner, V. Moliner, I.H. Williams, Phys. Chem. Chem.
G.S. Alagona, J. Profeta, P. Weiner, J. Am. Chem. Soc. 106
Carta a los Profesores Tenemos el honor y el placer de informarles que la 6ª edición del Concurso Internacional de Acordeón tendrá lugar los días 15 y 16 de octubre de 2011, en el Ayuntamiento de Roubaix. Esta gran manifestación artística, que no está vinculada con ninguna asociación ni referencia, se dirige a todos los acordeonistas a partir del 1er ciclo de aprendizaje, solista
PREPARATION FOR COLONOSCOPY WITH PREPOPIK The day before the examination: Drink only clear liquids (see back) all day from the time you get up, including at least one 32 oz bottle of Gatorade, to maintain a good state of hydration. You are not to eat any solids until after the colonoscopy. The evening before the examination: At 5 p.m. , take 1 pill of Dulcolax. At 7 PM, f

 Journal of Molecular Structure (Theochem) 632 (2003) 297–307
How important is the refinement of transition state structures
` ngels Gonza´lez-Lafont, Jose´ M. Lluch*
Departament de Quı´mica, Universitat Auto`noma de Barcelona, Bellaterra, Barcelona 08193, Spain
Received 30 October 2002; revised 18 December 2002; accepted 18 December 2002
In this paper the need to use a second derivatives direct algorithm to refine the location of transition state structures obtained
in enzymatic systems has been analyzed. The 25 approximate QM/MM transition state structures previously found by means ofa reaction coordinate approach for the three mechanisms of racemization of mandelate and propargylglycolate by mandelateracemase enzyme have been refined using a modified micro-iterative optimization method developed in this work. Therefinement of transition state structures is especially useful to assure that a structure, found as the highest potential energy pointon a profile depicted by a particular reaction coordinate, lies in the correct quadratic region. This is more important in thosesteps of the enzymatic process where the selected reaction coordinate may not reflect quite accurately the geometrical changestaking place in the active site.
Journal of Molecular Structure (Theochem) 632 (2003) 297–307
How important is the refinement of transition state structures
` ngels Gonza´lez-Lafont, Jose´ M. Lluch*
Departament de Quı´mica, Universitat Auto`noma de Barcelona, Bellaterra, Barcelona 08193, Spain
Received 30 October 2002; revised 18 December 2002; accepted 18 December 2002
In this paper the need to use a second derivatives direct algorithm to refine the location of transition state structures obtained
in enzymatic systems has been analyzed. The 25 approximate QM/MM transition state structures previously found by means ofa reaction coordinate approach for the three mechanisms of racemization of mandelate and propargylglycolate by mandelateracemase enzyme have been refined using a modified micro-iterative optimization method developed in this work. Therefinement of transition state structures is especially useful to assure that a structure, found as the highest potential energy pointon a profile depicted by a particular reaction coordinate, lies in the correct quadratic region. This is more important in thosesteps of the enzymatic process where the selected reaction coordinate may not reflect quite accurately the geometrical changestaking place in the active site.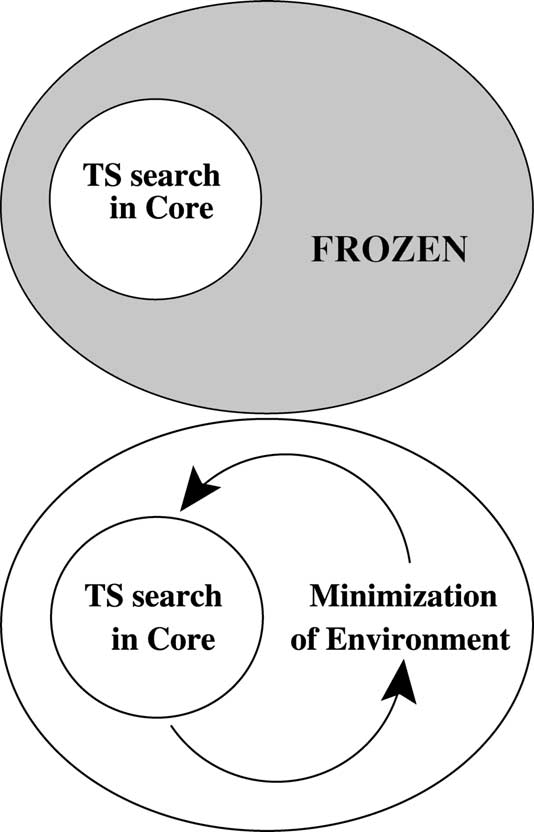 X. Prat-Resina et al. / Journal of Molecular Structure (Theochem) 632 (2003) 297–307
optimization with Cartesian coordinates will beconsidered.
X. Prat-Resina et al. / Journal of Molecular Structure (Theochem) 632 (2003) 297–307
optimization with Cartesian coordinates will beconsidered.
 X. Prat-Resina et al. / Journal of Molecular Structure (Theochem) 632 (2003) 297–307
the interaction between core and environment, andhow often the environment must be relaxed duringthe transition state structure search in the core. Allthese features will not be discussed here.
X. Prat-Resina et al. / Journal of Molecular Structure (Theochem) 632 (2003) 297–307
the interaction between core and environment, andhow often the environment must be relaxed duringthe transition state structure search in the core. Allthese features will not be discussed here.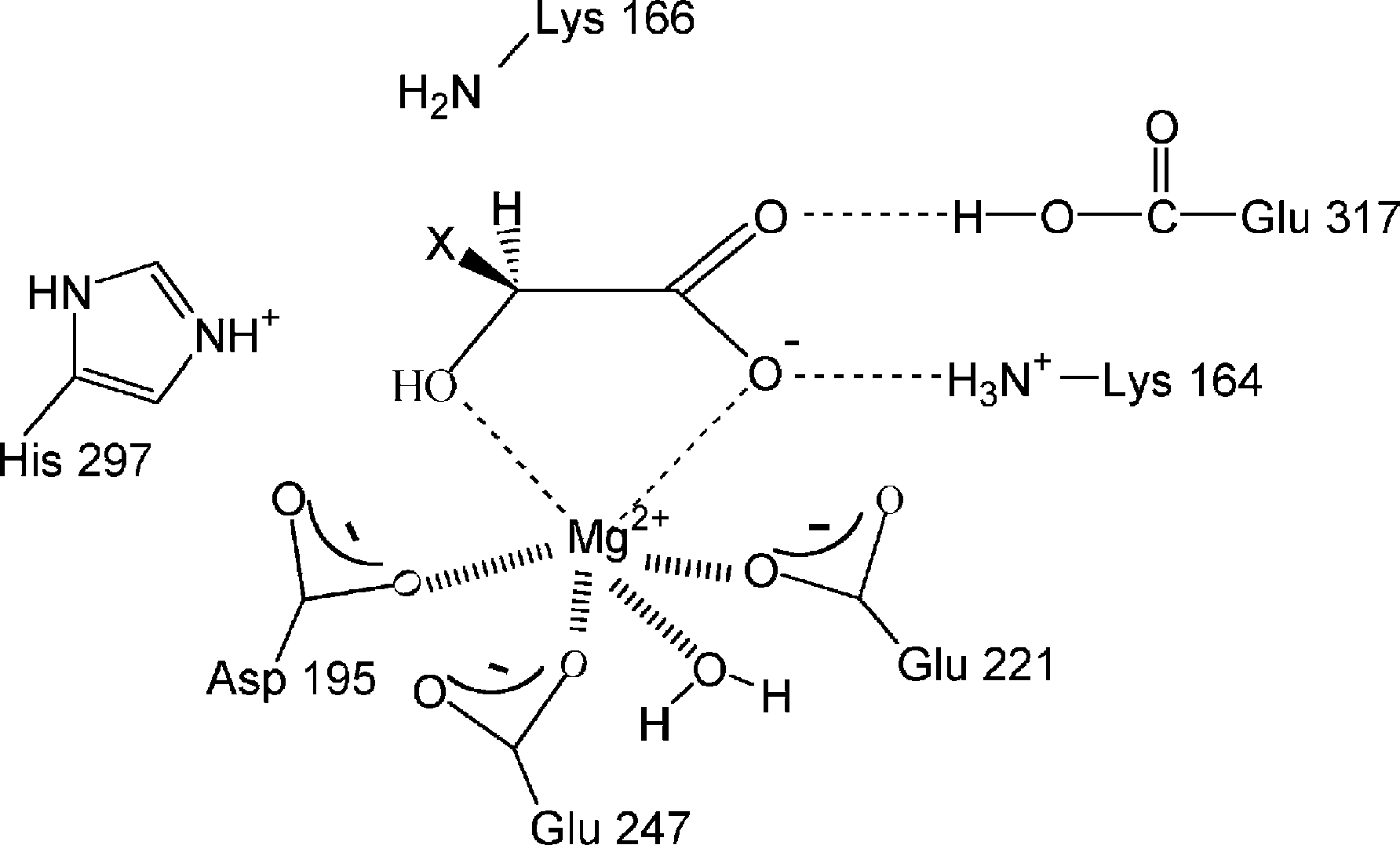
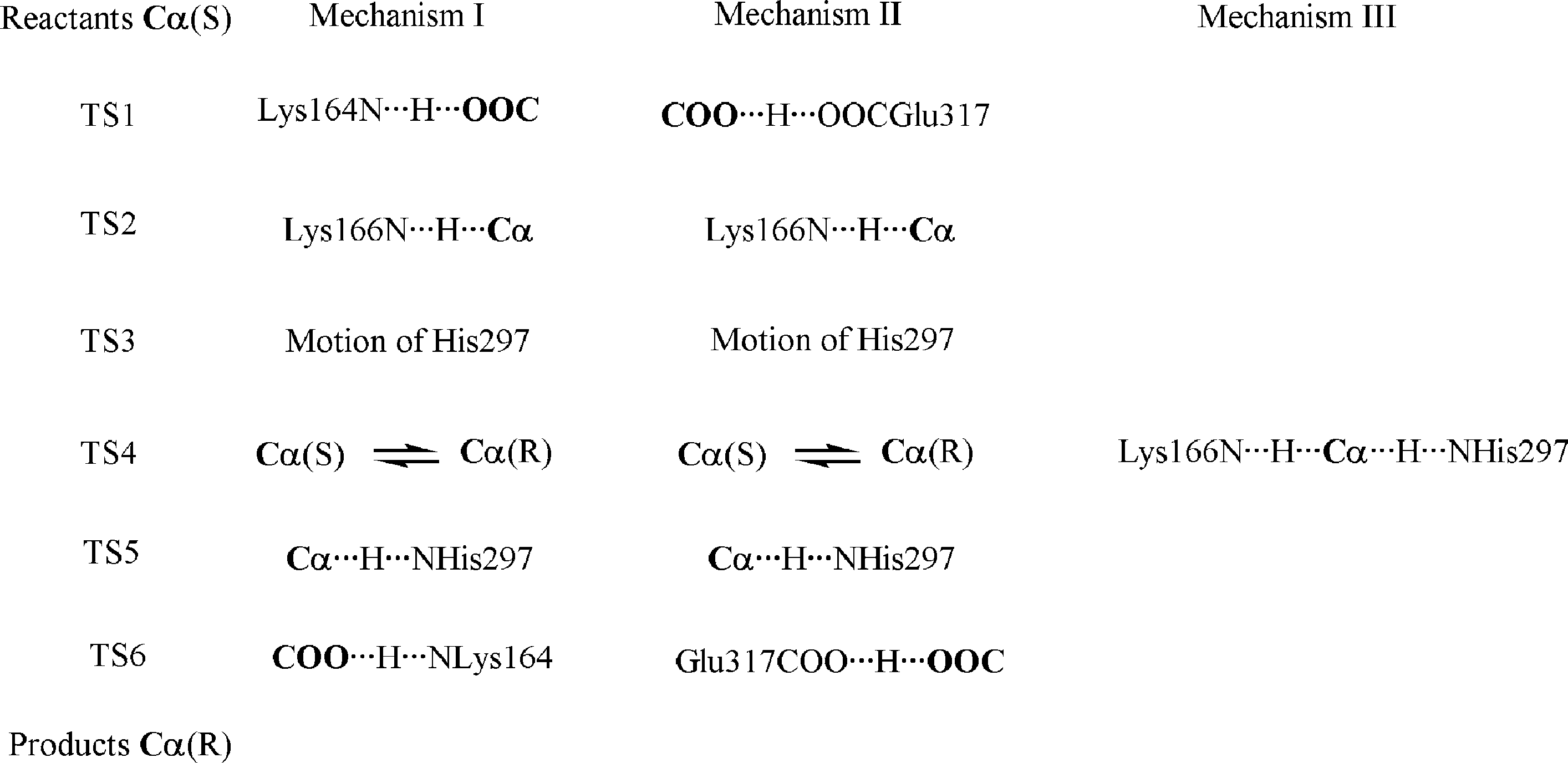 X. Prat-Resina et al. / Journal of Molecular Structure (Theochem) 632 (2003) 297–307
transfer neither from Lys164 nor Glu317 beingrequired.
X. Prat-Resina et al. / Journal of Molecular Structure (Theochem) 632 (2003) 297–307
transfer neither from Lys164 nor Glu317 beingrequired.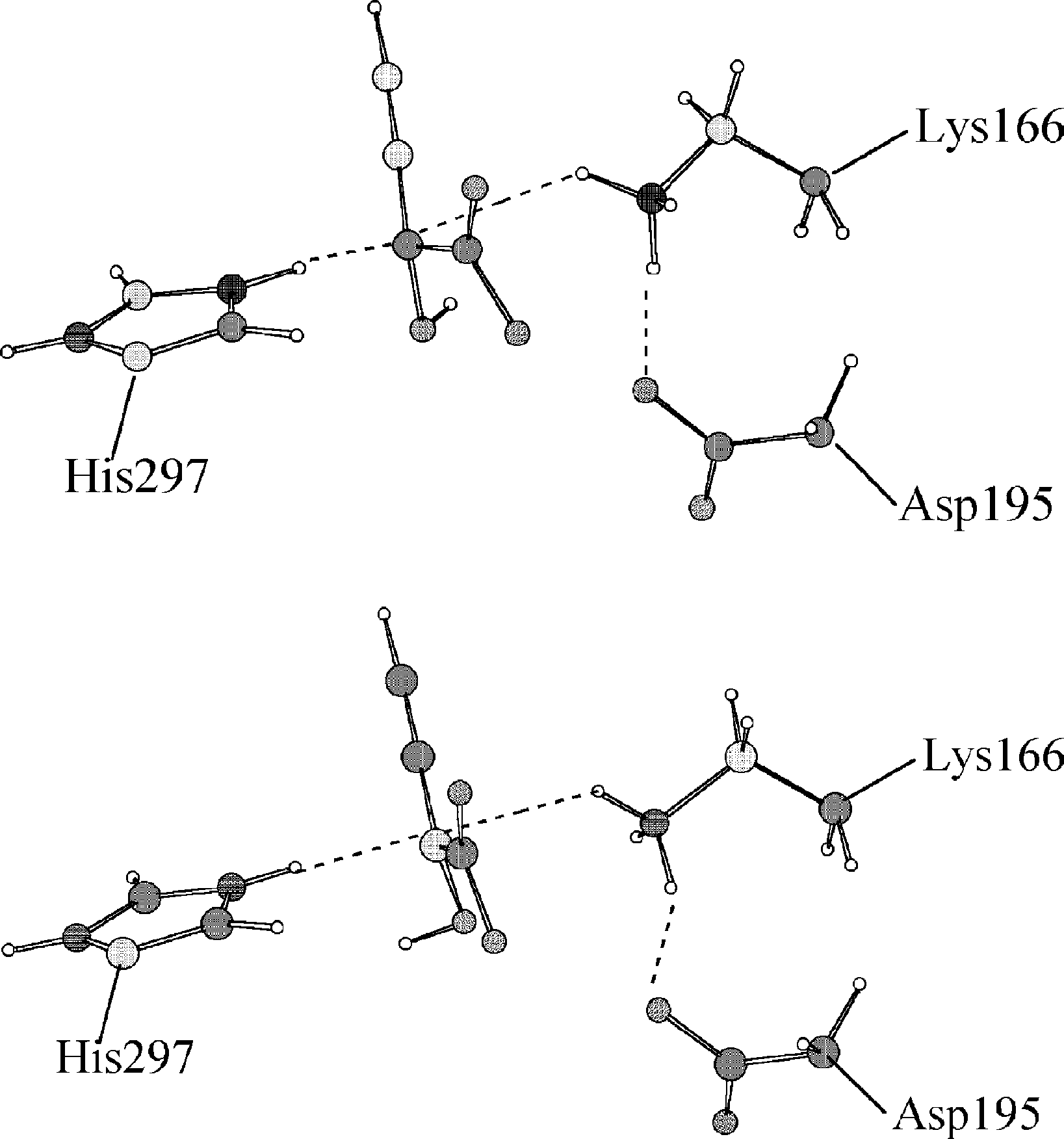 X. Prat-Resina et al. / Journal of Molecular Structure (Theochem) 632 (2003) 297–307
˚ ) for the transition state structures corresponding to the racemization of mandelate and propargylglycolate according to mechanism II
coordinate method was away from the quadratic
of the refinement of the coordinates and the potential
region with the suitable curvature corresponding to
energy of the located structure but with the problem of
the actual transition state structure of this step. So in
a transition state structure found by the reaction
this case we are not just dealing with the convenience
coordinate method that may not fulfill the adequate
˚ ) for the transition state structure corresponding to the
racemization of mandelate and propargylglycolate according tomechanism III
Fig. 6. Transition state structures located with the reaction coordinate
method (upper structure) and the micro-iterative method (lower structure)
for the step 4 in mechanism I of the substrate propargylglycolate.
X. Prat-Resina et al. / Journal of Molecular Structure (Theochem) 632 (2003) 297–307
˚ ) for the transition state structures corresponding to the racemization of mandelate and propargylglycolate according to mechanism II
coordinate method was away from the quadratic
of the refinement of the coordinates and the potential
region with the suitable curvature corresponding to
energy of the located structure but with the problem of
the actual transition state structure of this step. So in
a transition state structure found by the reaction
this case we are not just dealing with the convenience
coordinate method that may not fulfill the adequate
˚ ) for the transition state structure corresponding to the
racemization of mandelate and propargylglycolate according tomechanism III
Fig. 6. Transition state structures located with the reaction coordinate
method (upper structure) and the micro-iterative method (lower structure)
for the step 4 in mechanism I of the substrate propargylglycolate.