Tadalafil appartiene alla classe degli inibitori selettivi della fosfodiesterasi di tipo 5, con un profilo farmacocinetico caratterizzato da un’emivita terminale di circa diciotto ore. Dopo somministrazione orale viene assorbito rapidamente e raggiunge concentrazioni plasmatiche massime in due ore. La biotrasformazione avviene principalmente tramite CYP3A4 con formazione di metaboliti inattivi, escreti in prevalenza con le feci. L’elevato legame alle proteine plasmatiche (>90%) assicura una distribuzione stabile. Nei confronti delle altre molecole della stessa classe, cialis compresse italia è noto per la durata prolungata dell’attività farmacologica.
Farmaco.fip.org
Biowaiver Monographs for Immediate Release Solid OralDosage Forms: Furosemide
G.E. GRANERO,1 M.R. LONGHI,1 M.J. MORA,1 H.E. JUNGINGER,2 K.K. MIDHA,3 V.P. SHAH,4 S. STAVCHANSKY,5J.B. DRESSMAN,6 D.M. BARENDS7
1Chemical Sciences Faculty, Pharmacy Department, National University of Co´rdoba, Co´rdoba, Argentina
2Faculty of Pharmaceutical Sciences, Naresuan University, Phitsanulok, Thailand
3University of Saskatchewan, Saskatoon, Saskatchewan, Canada
4International Pharmaceutical Federation FIP, The Hague, the Netherlands
5Pharmaceutical Division, College of Pharmacy, University of Texas at Austin, Austin, Texas
6Institute of Pharmaceutical Technology, J.W. Goethe University, Frankfurt, Germany
7RIVM—National Institute for Public Health and the Environment, Bilthoven, the Netherlands
Received 29 July 2009; accepted 29 October 2009
Published online 3 December 2009 in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/jps.22030
ABSTRACT: Literature and new experimental data relevant to the decision to allow a waiver ofin vivo bioequivalence (BE) testing for the approval of immediate release (IR) solid oral dosageforms containing furosemide are reviewed. The available data on solubility, oral absorption, andpermeability are sufficiently conclusive to classify furosemide into Class IV of the Biopharma-ceutics Classification System (BCS). Furosemide’s therapeutic use and therapeutic index, itspharmacokinetic properties, data related to the possibility of excipient interactions and reportedBE/bioavailability (BA) problems are also taken into consideration. In view of the data available,it is concluded that the biowaiver procedure cannot be justified for either the registration of newmultisource drug products or major postapproval changes (variations) to existing drug products. ß 2009 Wiley-Liss, Inc. and the American Pharmacists Association J Pharm Sci 99:2544–2556, 2010Keywords:
furosemide; absorption; bioequivalence; Biopharmaceutics Classification System
(BCS); permeability; solubility; regulatory science
new multisource products, are evaluated underconsideration of its biopharmaceutical and clinical
A biowaiver monograph of furosemide based on
properties. This evaluation refers to drug products
literature data, together with additional experimen-
containing furosemide as the single active pharma-
tal data, is presented. The risks of basing a
ceutical ingredient (API). The purpose and scope of
bioequivalence (BE) assessment on in vitro rather
this series of monographs have been previously
than in vivo study results for the approval of new IR
discussed.1 Summarized in few words, the aim is to
solid oral dosage forms containing furosemide (‘‘bio-
evaluate all pertinent data available from literature
waiving’’), including both reformulated products and
sources for a given API to assess the risks associatedwith a biowaiver. For these purposes, risk is definedin terms of the probability of an incorrect biowaiver
A project of the International Pharmaceutical Federation FIP,
decision as well as the consequences of an incorrect
Special Interest Group BCS and Biowaiver, www.fip.org/bcs.
decision in terms of public health and individual
This article reflects the scientific opinion of the authors and not
the policies of regulating agencies, the International Pharmaceu-
patient risks. On the basis of these considerations,
tical Federation (FIP) and the World Health Organization (WHO).
a recommendation can be made as to whether a
Correspondence to: D.M. Barends (Telephone: 31-30-2744209;
biowaiver is advisable or not. It is pointed out that
Fax: 31-30-2744462; E-mail: [email protected])
these monographs do not simply apply the various
Journal of Pharmaceutical Sciences, Vol. 99, 2544–2556 (2010)ß 2009 Wiley-Liss, Inc. and the American Pharmacists Association
regulatory documents, but also serve as a critical
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 6, JUNE 2010
evaluation of these documents. Biowaiver mono-
headache, hypotension, muscle cramps, dry mouth,
graphs have already been published for acetamino-
thirst, weakness, etc.20 There is generally no need to
phen (INN: paracetamol),2 acetazolamide,3 aciclovir,4
amitriptyline,5 atenolol,1 chloroquine,6 cimetidine,7diclofenac,8 doxycyline hyclate,9 ethambutol,10 ibu-
profen,11 isoniazid,12 metoclopramide,13 predniso-lone,14 prednisone,15 pyrazinamide,16 propranolol,1
quinidine,17 ranitidine,18 rifampicin,19 and verapa-mil.1 They are also available on-line at www.fip.
The aqueous solubility of furosemide at room
temperature has been reported to be 0.01825 mg/mL.21 Its aqueous solubility increases as function ofthe pH of the medium from 0.18 mg/mL at pH 2.3 to
13.36 mg/mL at pH 10.22 Martindale reports thatfurosemide is practically insoluble in water, corre-
profile of furosemide at 308C showed a minimum of
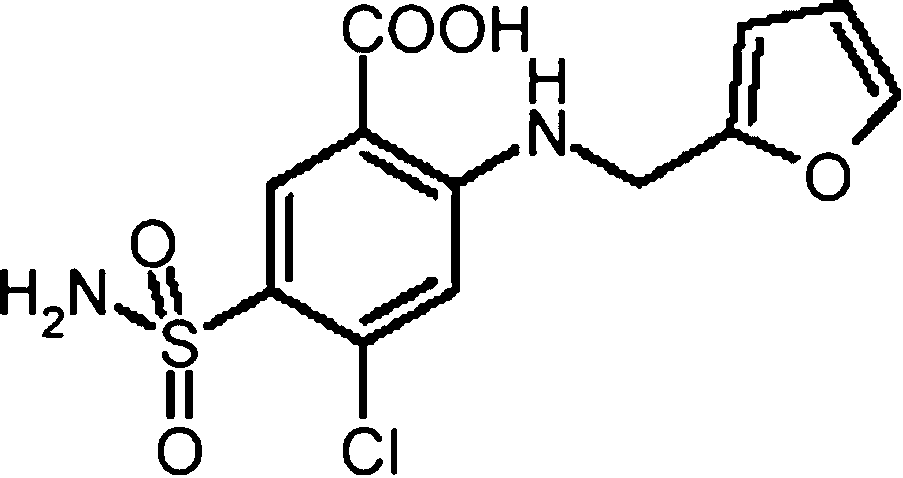
Chemical name: 4-chloro-N-furfuryl-5-sulphamoy-
0.010 mg/mL at pH 2.0 and a maximum of 21.9 mg/mL
lanthranilic acid or 5-(aminosulfonyl)-4-chloro-2[(2-
at pH 8.0, followed by a marginal decrease to about
furanylmethyl)amino] benzoic acid. Its structure is
18 mg/mL above pH 8.0.23 Other workers reported a
saturation solubility at pH 4.6 and 378C of 0.008 mg/mL.24 The equilibrium solubility of furosemide at
Therapeutic Indication, Therapeutic Index, and Toxicity
378C in Krebs Ringer buffer at pH 5.0 was 0.33 mg/mL,increasing to $1.5 mg/mL at pH 6.5 and 1.9 mg/mL
Furosemide is a loop diuretic that is used orally in the
at pH 7.4.25 New solubility data at pH values within
treatment of edematous states associated with
the ranges required by the various Guidances26–29
cardiac, renal, and hepatic failure and the treatment
were measured1 in triplicate at pH 1.0; 2.8; 3.8; 4.8;
and 7.5 using the standard USP shake-flask method,
The usual dosage is 40–120 mg/day. For the
with stirring for 48 h at 378C. A summary of the
treatment of mild cases of edema, doses as low as
literature data as well as the new data are presented
20 mg can be effective, whereas for severe cases of
in Table 1. No data on the stability of furosemide in
edema doses as high as 600 mg/day may be required.20
human gastric and intestinal fluids were found in the
For the treatment of chronic renal impairment the
dose can be as high as 1.5 g/24 h. Furosemide inhibitsthe reabsorption of sodium and chloride in the
ascending limb of the loop of Henle and also in the
A furosemide sodium salt is known, but is used only in
early distal tubules. Excretion of sodium, potassium,
parenterals, such as furosemide for injection USP.30
calcium, and chloride ions is increased and water
Seven polymorphic forms are known: four true
excretion enhanced.20 Most adverse effects of furose-
polymorphs (I, II, III, IV), two solvates (IV—DMS
mide occur at high doses and/or prolonged use.
and V—dioxane) and one amorphous form,31–34 but
Serious effects are uncommon, the most common
polymorph-dependent bioavailability (BA) has not
being fluid and electrolyte imbalance, including
been reported to date in the literature.
hyponatraemia, hypokalaemia, and hypochloraemicalkalosis. Signs of electrolyte imbalance include
Furosemide is a weak acid with an acidic pKa value of3.8 (carboxylic acid).35
Log P (n-octanol/water) values of 2.2935 and 1.8136have
at pH values of 7.39, 5.86, and 2.58 have beenreported to be À1.20, À0.10, and 1.78, respectively.36A log D (pH 7.4) value of À0.69 has been measured.35Kasim et al.37 calculated log n-octanol/water partition
1Experiments performed at the Pharmacy Department, Chemi-
cal Sciences Faculty, National University of Co´rdoba, Argentina.
The pH was measured after the addition of the drug.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 6, JUNE 2010
Table 1. Solubility (mg/mL) of Furosemide: Literature Data and New Experimental Data. Also shown: the CorrespondingDose/Solubility Ratio (D/S) (mL) at 378C for three tablet strengths
aCalculated from the solubility data at 378C; the critical limit is 250 mL.26–29
bStrength on WHO essential medicines list.38
cA D/S value exceeding the critical limit.
coefficients for furosemide using two different
tions, Lasix1 and Furix1, each in a dose of 40 mg, and
methods, finding values of 1.9 and 0.74, respectively.
also after intravenous (i.v.) administration in eight
For metoprolol, following the same methodologies,
healthy subjects. Absolute BA was reported to be 56%
the authors reported values of 1.35 and 1.72,
for Lasix1 and 55% for Furix1, with a range of 20–
84% between individuals and 20–61% within anindividual, indicating extensive variability after
oral administration. Extensive variability was also
The WHO recommended oral dosage form strength is
observed in mean absorption time and urinary
40 mg.38 Table 2 shows IR furosemide tablets with a
excretion. The intra-subject variability was thought
marketing authorization (MA) in Germany (DE),39
to depend mainly on the absorption process, since
Denmark (DK),40 Finland (FI),41 France (FR),42 The
repeated i.v. doses showed only marginal intra-
Netherlands (NL),43 Norway (NO),44 Spain (ES),45
subject variability, but, as this study was severely
Sweden (SE),46 United Kingdom (UK),47 and the
underpowered, it is not possible to draw robust
United States (US).48 These MAs cover a very wide
range of strengths: from 20 mg up to 500 mg.
The hypothesis that furosemide exhibits site-
specific absorption was investigated in the rat model. In this animal model, Chungi et al.54 reported
absorption to be biexponential and rapid whenadministered to the stomach but slower when
administered to the small intestine. The most rapid
Furosemide is fairly rapidly absorbed from the
absorption occurred after administration to the
gastrointestinal (GI) tract. Its BA was reported to
stomach at a pH of 3. In man, the absorption of
be about 60–70%, but the absorption is variable and
furosemide is also site-specific and takes place
erratic.20 Others report a poorer oral BA of 50%49–51
primarily in the upper parts of the small intestine.
or 37–51%.52 Peak serum concentrations (Cmax) occur
Clear et al.55 released furosemide using an Intelisite1
between 60 and 90 min with concentrations falling
capsule at specific sites in the GI tract, finding that
below the limit of detection between 3 and 4 h after
the absorption window of furosemide in the upper GI
ingestion. The rate and extent of absorption show
tract is narrow: when drug release took place in the
large inter- and intra-subject variabilities. Absorp-
proximal intestine instead of the stomach, the area
tion following oral administration is influenced by the
under the concentration time curve (AUC) for
dosage form, underlying disease processes, and by
furosemide decreased markedly, by 29%. From these
the presence of food.50 Grahne´n et al.53 investigated
studies it was concluded that, in humans, furosemide
the intra-subject variation in BA with respect to rate
is most rapidly absorbed from the upper GI tract
and extent of absorption between two tablet formula-
following dissolution in the stomach.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 6, JUNE 2010
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 6, JUNE 2010
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 6, JUNE 2010
Absorptive behavior differences were reported
The pharmacokinetics of furosemide are reported to
between dosage forms. Hammarlund et al.51 studied
be linear over the oral dosage range of 20–80 mg.61
in 8 subjects the mean time for the different steps in
Furosemide plasma profiles often exhibit secondary
absorption for i.v. and different modes oral adminis-
or multiple peaks following either oral or i.v.
tration of furosemide. The mean absorption times for
administration.52,62,63 These phenomena have been
all oral doses were significantly longer than the mean
attributed to enterohepatic cycling of the drug.50
absorption times after i.v. administration, indicating
However, furosemide is mainly excreted in the urine,
absorption rate-limited kinetics. Absorptive behavior
largely unchanged. There is some excretion via the
differences were also reported between solutions
bile and nonrenal elimination, but the small amount
versus tablets. Waller et al.56studied two furosemide
of furosemide reabsorbed after biliary elimination is
tablets and an aqueous solution in 21 healthy adult
not sufficient to account for the secondary peaks.20
males. The peak plasma furosemide concentration
Other authors explain the multiple plasma peaks
obtained with the solution was significantly greater
with an erratic absorption behavior.64 However, this
than with the tablet formulations. Also, the time to
hypothesis is not consistent with multiple peaks after
peak occurred significantly earlier with the solution.
This finding was confirmed by McNamara et al.57
evaluating the relative BA of five tablets and an oralsolution in 12 normal volunteers in a crossover
Several authors report permeability data of furose-
design; compared to the solution, all tablets formula-
mide; 25,65–72 they are shown in Table 3. Furosemide
tions exhibited lower peak furosemide concentration.
is a known substrate of efflux transporters.65 Motz25
Absorptive behavior differences were also reported
applied a proton gradient between an apical to
between the fasting and nonfasting state. In the study
basolateral compartment (A-B) transport study with
of Hammarlund et al.,51 food delayed the absorption
A ¼ pH 6.5 and B ¼ pH 7.4, respectively, resulting in a
on average by 60 min. When a 40 or 80 mg tablet of
flux–efflux high ratio of $50. The large directional
furosemide was taken orally by healthy adults in the
differences in transport rates in the Caco-2 cells have
fasting state, a detectable concentration of drug
been attributed to the secretion of this API by efflux
appears in the serum within 10 min and peaks
systems such as the P-glycoproteins on the one hand
between 60 and 90 min at a level of 1–3 mg/mL,58,59
and to a significant paracellular contribution to
but when taken in close proximity to a meal, there is a
delay in its appearance in plasma, and a lower peak
It has been reported that apparent permeability
concentration of about 1 mg/mL was reported after
( Papp) of furosemide can be affected by the presence of
$2 h.60 Despite the difference in peak serum con-
the excipient Tween-801 (polysorbate 80). Rege
centrations the total amount of furosemide absorbed
et al.70 reported an increase in the apical-to-baso-
is similar.60 Kelly et al.52 also found that postprandial
lateral (A-B) transport of furosemide in the presence
administration of furosemide results in delayed
of Tween-801, which neutralized the asymmetry in
appearance of the drug in serum, lowered Cmax and
transport. Polysorbate 80 is a known P-glycoprotein
more prolonged concentrations. Beermann and Mid-
skov60 reported a reduced but parallel plasma
effect, observing not only an increase in Papp (A-B)
concentration versus time profile between the fasting
but also a decrease in Papp basolateral-to-apical (B-A)
in a Caco-2 cell model. Motz25 also found that vitamin
When metoprolol was included as a reference, its permeability is also reported.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 6, JUNE 2010
E d-alpha-tocopheryl poly(ethylene glycol)succinate,
different brands of 40 mg furosemide tablets available
another P-glycoprotein inhibitor,73 increased Papp
in Thailand was evaluated. Only four brands passed
(A-B) of furosemide, while Papp(B-A) was reduced.
the specification for dissolution (apparatus 2 at50 rpm in pH 7.4 phosphate buffer). The original
Distribution, Metabolism, and Elimination
brand (brand A) and the three local brands (brand B,
Furosemide is up to 99% bound to plasma proteins.30
C, and D) which showed differences in dissolution
The clearance of furosemide is generally reported to
characteristics were selected for a BA study in eight
be in the range of 0.09–0.18 L/h/kg. The half-life of
healthy subjects. Plasma furosemide concentrations
furosemide is in the range of 30–120 min and it is
and urine output, and sodium, chloride, and potas-
mainly excreted in the urine, largely unchanged.20 In
sium excretion were measured. The relative BA of
end-stage renal disease the half-life may reach almost
furosemide with respect to brand A was 70% (brand
10 h and in neonates the half-life is also prolonged,
B), 113% (brand C), and 95% (brand D); these
since renal function is not yet mature at birth. As well
differences were deemed not statistically significant
as renal elimination there is also some excretion
different at a 95% confidence level, but certainly at
via the bile, with the role of nonrenal elimination
least brand A would not have met the current 0.80–
considerably greater in renal impairment.50
1.25 criterion for the AUC. As the clinical response in
Furosemide has two metabolites, furosemide glu-
terms of diuresis and electrolyte excretion between
the four brands was not significantly different at a95% confidence level, the authors concluded that theformulations were clinically equivalent. Here again,
the power of the study was likely too weak toappropriately detect differences.
Excipients and/or Manufacturing Variations
Awad et al.80 estimated the BE of Diusemide versus
Reports of BE studies between furosemide IR drug
Lasix, each containing 40 mg of furosemide, in 20
products show inconsistent results.74–78 However,
healthy volunteers. The compositions of the products
most of these studies were carried out 20–30 years
were not reported. No significant differences were
ago, when BE was not defined according to the
found in AUC, Cmax, tmax, cumulative urine volume,
current biostatistical standards. Nowadays drug
products are considered bioequivalent if, with high
Although this analysis led the authors to conclude
probability, the hypothesis that two formulations
to BE between the two products, the power of the
are bioinequivalent can be rejected,26–29 whereas at
study was undoubtedly too weak to conclude that the
the time most of the literature studies were con-
products were bioequivalent using today’s BE stan-
ducted, two formulations were considered bioequiva-
lent if no significant differences in pharmacokinetic
Nakib et al.81 reported BE of a brand of furosemide
parameters were observed. As a result, in several
40 mg tablets versus Lasix1. The compositions of the
early studies formulations were reported to be
products were not reported. The study included 24
bioequivalent, even though by current biostatistical
fasting, healthy, male volunteers and 90% confidence
intervals of the ratios of AUC0–t, AUC0–1, and Cmax of
met BE criteria due to insufficient power in the
the two formulations were within the 80–125% range.
study design. Additionally, most references do not
Applying current BE standards, the two products
report results in sufficient detail to allow recalcula-
tion of the data according to current biostatistical
Cuadrado et al.82 studied two 40 mg furosemide
formulations versus a reference, the identity of which
not was revealed. The compositions of the products
marketed brands of furosemide, Impugan1 (A/S
were also not reported. The study included 24 healthy
Dumex, Copenhagen, DK) and Lasix1 (Hoechst
volunteers; plasma furosemide concentrations, urine
AG, Frankfurt (M), DE), in five healthy volunteers.
output and sodium, chloride, and potassium excre-
The compositions of the products were not reported.
tions were measured. AUC0–1, and Cmax and were
Although time of the peak levels, AUC and the
tested for BE after ln-transformation and ratios of
urinary recovery after the oral administration did not
tmax were evaluated nonparametrically. Ninety per-
differ significantly using Student’s paired t-test, the
cent confidence intervals for AUC0–1 were 0.94–1.19;
power of the study was undoubtedly too weak to
for Cmax 0.96–1.31 and for tmax 0.55–1.00, respectively
conclude that the two products were bioequivalent.
and BE between both formulations was concluded by
A Thai study79 compared the in vitro dissolution
the authors for all parameters except for tmax. The
and clinical response among marketed furosemide
methodology used was in line with current standards,
drug products. The compositions of the products were
but many regulatory authorities would conclude that
not reported. The in vitro dissolution of thirteen
these formulations do not meet current BE criteria,
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 6, JUNE 2010
since for Cmax an acceptance criteria of 0.8–1.25 is
Rubinstein and Rughani85 studied furosemide
40 mg tablets, prepared with four different binders:
Grahne´n et al.53 reported a study of two tablet
polyvinylpyrrolidone, starch mucilage, stearic acid,
formulations, Lasix1 and Furix1, each at a dose of
and methylhydroxyethyl cellulose. BA was assessed
40 mg, in eight healthy subjects. For these subjects
in four healthy males with reference to an oral
the products were demmed nonequivalent, based
solution. The tablets containing polyvinylpyrrolidone
on AUC: However, after extending the study to
and methylhydroxyethyl cellulose showed point
16 subjects, the authors considered the products to be
estimate of relative BA values of 72% and 72%,
bioequivalent, based on a <6% probability that there
respectively, while the starch mucilage formulation
was a >20% difference in AUC. This criterion does not
and the stearic acid formulation showed relative BA
values of 54% and 35%, respectively. This consider-
Studies suggesting bioinequivalence2 between fur-
able decrease of the BA of furosemide by starch and
osemide tablets have also been reported. Wolf-
stearic acid was not confirmed by other excipient
Coporda et al.84 evaluated two oral preparations of
interaction data, see below. As this study was
furosemide, a Croatian test product and the reference
severely underpowered, it is not possible to draw
preparation, Lasix1 (Hoechst AG), at a dose of 500 mg
any robust conclusions from the data.
in 15 healthy male volunteers. The compositions
Table 2 shows the excipients present in IR
of the products were not reported. The test product
furosemide tablets with an MA in various countries.
showed a considerably higher Cmax; statistically
As over the years the criteria for BE have been
significant shorter tmax and significantly higher
changed, it cannot be assumed that all these drug
AUC than the reference preparation. The relative
products successfully had passed an in vivo BE study
BA of the test product was 129% and thus not
that would be in conformity with the present
equivalent to the reference according to current BE
regulations. However, in view of their MA, there
standards. These products maybe even bioinequiva-
can be little doubt on their clinically efficacy and
lent, in view of the large differences in the point
safety. It can therefore be argued that the excipients
estimates of these pharmacokinetic parameters. The
present in these drug products do not exert a
brand with the highest Cmax also showed the fastest
significant effect on the rate and extent of absorption
in vitro dissolution but the test conditions used were
of furosemide. Table 2 includes polysorbate 80, an
excipient which showed an effect on the Caco-2
McNamara et al.57 evaluated three brands of 40 mg
permeability of furosemide.70 This suggests that the
furosemide tablets, one of which was Lasix1, and one
Caco-2 excipient interaction studies may have over-
oral solution in 12 volunteers. For two brands,
discriminated. Table 2 also includes starch and
including Lasix1, two different batches were included
stearic acid, excipients which were reported by
in the study, one of which was designated as
Rubinstein and Rughani to decrease the BA of
reference. The compositions of the formulations were
furosemide considerably is possible that the amounts
not reported. Plasma and cumulative urine concen-
used were quite different between the test formula-
trations of furosemide were measured, as well as the
tions and those described in Table 2.
in vitro dissolution of the tablets in USP Apparatus II
Dissolution and In Vitro–In Vivo Correlations (IVIVCs)
(paddle) at 50 rpm in acetate buffer pH 4.6 and 5.6. With respect to the usual pharmacokinetic para-
The USP 32 specification for dissolution of furosemide
meters, all tablet formulations were significantly
tablets is not <80% (Q) dissolved in 60 min in 900 mL
different from the reference at the 95% confidence
of pH 5.8 phosphate buffer, using the paddle
level, with point estimate BA ranging from 70% to
91%. This study also reported a wide intra-subject
There are some reports describing successful
variability from oral dosage forms. Applying current
IVIVCs for furosemide drug products.82–86 Rubinstein
BE standards, it would most probably be concluded
and Rughani85 reported that the observed differences
that all tested tablet formulations failed to meet the
in BA of furosemide tablets with different exci-
BE criteria; some of these formulations might even be
pients were reflected in in vitro drug release in
distilled water. Stu¨ber et al.86 studied the BA of sixcommercial tablet preparations in six volunteers. Theidentities and the composition of the tested tabletswere not reported. One tablet reached only 80% of the
AUC of the reference tablet; its Cmax was lower and its
Bioinequivalence implies that the regulatory defined confidence
interval of one, or more, BE attributes (AUC, Cmax, Tmax) falls fully
tmax longer. The tablet with the lowest BA in term of
outside of their regulatory acceptance range, whereas failure to
AUC, Cmax, and tmax also showed lower in vitro
meet BE criteria implies that the regulatory defined confidence
dissolution than the reference tablet in each of four
interval of one, or more, BE attributes does not fully fall inside theirregulatory acceptance range.83
different methods: pH 7.8/paddle 25 rpm; pH 7.8/
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 6, JUNE 2010
paddle 50 rpm; pH 5.3/paddle 50 rpm and flow-
through cell/pH 7.8. The difference in vitro dissolu-tion was most pronounced at pH 5.3/paddle 50 rpm;
under this condition the time needed to reach 50%
Solubility criteria defined in present regulatory
dissolution for the tablet with the low BA was 4.4
guidances26–29 for classifying an API as highly soluble
require the highest dosage strength to be soluble at
Investigating the dissolution of two brands of
378C in 250 mL aqueous solution over the pH range of
furosemide tablets Prasad et al.87 found negligible
1.0–6.8, according to the EU28,29 and WHO26 gui-
differences at pHs higher than 4.6; the brand
dances, or 1.0–7.5 according to the FDA guidance.27
dissolving poorly at pH 4.6 also had an inferior BA
The dose to solubility ratio (D/S) at 378C of the most
with respect to both Cmax and AUC. In a study of four
often used strength, 40 mg, exceeds the critical value
commercial and two experimental furosemide tablets
of 250 mL at pH 4.8 and below; the 500 mg tablet
Kingsford et al.88 reported a good correlation between
exceeds the critical D/S value at pH 5.0 and below, see
the percentage dissolved in 30 min in buffer pH 5.0 at
Table 1. Hence, furosemide is not highly soluble.
378C in the rotating basket and the percentage offurosemide recovered in the urine.
McNamara et al.57 tested five lots of furosemide
tablets for dissolution in acetate buffer at pH values of
The FDA defines highly permeable as having a
4.6 and 5.6, using a USP paddle apparatus at 50 rpm
fraction dose absorbed of not <90%.27 The WHO
at pH 4.6 and 5.6. The products dissolved faster
Guideline set a limit of not <85% of the fraction dose
and more completely at pH 5.6. Correlations of
absorbed.90 The EMEA Note for Guidance presently
mean in vivo parameters with in vitro dissolution
in force is less precise, stating that ‘‘linear and
approached statistical significance, with a somewhat
complete absorption indicating high permeability
higher correlation with the parameters derived from
reduces the possibility of an IR dosage form influen-
dissolution at pH 4.6 than at pH 5.6. However, since
cing the BA.’’28 The draft revision to that Guidance
two products showing only small differences in
states that an extent of absorption !85% is generally
differences in dissolution at pH 4.6, the authors
Furosemide is incompletely absorbed after oral
concluded that this medium was overly discriminat-
administration to healthy subjects and also in
ing and that the pH 5.6 medium would be more
patients with various diseases.59,91 Additionally,
appropriate for assuring batch uniformity and BE of
Attachment A of the FDA Guidance classifies
Waller et al.56 compared tablets in vivo and in vitro
Caco-2 data are in line with that classification. For
of identical composition but produced by a slightly
drug transport in Caco-2 monolayers, a cutoff point
different manufacturing method. The in vivo study in
for highly permeable APIs of Papp ¼ 10À5 cm/s, was
21 healthy human volunteers showed the relative
proposed to ensure a fraction dose absorbed higher
BAs of two tablets to be 89% and 101% compared to
than 95%.92 Similarly, a cutoff limit of Papp from
the solution, respectively, as determined by AUC, and
2 Â 10À6 to 10À5 cm/s as a boundary of highly perme-
these were reported to be not different at a 95%
able were proposed by Rinaki et al.93 Other workers
confidence level. After 30 min dissolution testing
proposed that a cutoff limit of Papp of 2 Â 10À6 cm/s in
at pH 5.8 in the paddle apparatus operated at
Caco-2 is commensurate with 100% absorption.94 The
50 rpm, one tablet showed 83% dissolution, the other
apical-to-basolateral, that is, the absorptive Papp
tablet 49% dissolution. Under the same conditions,
values reported for furosemide, being in the range
but using a medium composed at pH 4.6, the
of 0.1–0.5 Â 10À6 cm/s, are a factor of 4–20 below these
same tablets released 41% and 17%, respectively.
boundary values. It can be questioned if absolute
Since the two products show only small differences
Caco-2 permeability data are not so laboratory
in pharmacokinetic parameters, but marked differ-
specific that a general limit cannot be set. However,
ences in dissolution at pH 5.8, and even larger
Table 3 shows that in all studies where metoprolol
differences at pH 4.6, this study suggested that
was included as a reference, the permeability of
dissolution testing of furosemide tablets tends to
furosemide was far lower than the permeability of
be overly discriminating, particularly at pH 4.6.
metoprolol; metoprolol is the reference substance in
Qureshi and McGilveray89 reported a collaborative
classifying any other substance as highly permeable
study on the in vitro dissolution of 40-mg furosemide
or not highly permeable. And the log P and C log P
tablets in buffer pH 5.8 and buffer at pH 4.6. About
values are likewise in line with the classification of
20–38% of the variability in dissolution was not
furosemide as not highly permeable, although corre-
product related but came from the dissolution test
lations of log P values with human intestinal perme-
ability show both false positives and negatives.37
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 6, JUNE 2010
Patient’s Risks Associated With Nonequivalence
The most recent WHO Guideline,90 as well as Kasim
The regulations of the FDA, the WHO, and also the
et al.37 and Lindenberg et al.,95 all classify furosemide
draft Guideline on BE of the EU exclude Narrow
as Biopharmaceutics Classification System (BCS)
Class IV. Moreover, Wu and Benet96 classified
ing.26,27,29 The therapeutic plasma concentration
furosemide as Class IV in their Biopharmaceutics
for furosemide ranges from 1 to 6 mg/mL, with toxicity
Drug Disposition Classification System (BDDCS), a
occurring in the range 25–30 mg/mL.97 According to
system using the disposition characteristics of an API
the FDA definition of NTI,98 furosemide is not a NTI
as an estimate of its GI permeability. On the basis of
drug, since there is more than a twofold difference
literature data presented in this monograph, new
between the minimum toxic concentration (25 mg/mL)
data generated, as well as the classification of
and the minimum effective plasma concentration
furosemide by other groups, it can be concluded that
(1 mg/mL). The Pan American Health Organization
PAHO classified furosemide as an intermediatehealth-risk drug in view of the margin between the
Risk of Nonequivalence Caused by Excipients
nontoxic maximum and effective minimum concen-
trations and its adverse effects. This organizationclassified furosemide as having an intermediate
Many studies reported in the literature asserted that
probability of a minor complication of the disease
the drug products studied were bioequivalent, but
and/or mild adverse reactions at plasma concentra-
most studies used small subject numbers and
tions outside the therapeutic window of the drug.99
statistical methods that do not meet current require-
The current EU regulation does not mention the
ments. Only the Nakib et al.81 and Cuadrado et al.82
concept of NTI, but states that noncritical therapeutic
studies appeared to have reached a BE conclusion
range should be considered, defined as requirements
based on currently accepted methodology. On the
of special precautions with respect to precision and
other hand, in the Thai,79 Wolf-Coporda et al.,84
accuracy of dosing, for example, the need for critical
McNamara et al.,57 and Rubinstein and Rughani85
studies, at least one of the products showed such large
Furosemide is used for serious indications such as
differences in pharmacokinetic parameters from
cardiac insufficiency and pulmonary hypertension. In
those of the comparator that it is most likely that
many therapeutic situations, including edema of
that product would be declared bioinequivalent after
varying severity and oliguria, dose titration in the
application of statistical testing. Although most of
individual patient is recommended.20 This is partly
these products were exploratory or test formulations,
because furosemide shows large intra- and inter-
the results indicate that changes in composition and/
subject variabilities in BA after oral administration
or variations in manufacturing techniques can indeed
and partly due to variability in patient response to
have an impact on the BA of furosemide.
furosemide. Although a bioinequivalence betweentwo furosemide drug products could easily bemasked
Surrogate Techniques for In Vivo BE Testing
by the large intra- and inter-subject variabilities inBA and the dose titration, approving drug products
A variety of dissolution test conditions have been used
which cannot meet BE criteria is not an option for
to link in vitro to in vivo performance. In general, the
results indicate that dissolution in media with a pH inthe range of pH 5.0 to 5.8, that is, the pH of the USPdissolution test, will detect differences in BA. Testing
in more acidic dissolution media, such as pH 4.6,tends to be overly discriminating, whereas tests in
Furosemide is BCS Class IV, so both the in vivo
more alkaline media, such as pH 7.8, tend to lose
dissolution and the in vivo permeability can be critical
discrimatory power. To date, however, there are not
to in vivo performance of oral furosemide drug
enough data with any one set of in vitro test
products. No data are available in the literature
conditions to allow a firm conclusion on its reliability
about its stability in human gastric and intestinal
as a predictor for in vivo performance.
fluids. Likewise, no surrogate methods have been
Further, there are hints in the literature data that
identified in the literature that would reliably
in vitro permeability of furosemide may show an
forecast the in vivo performance of oral furosemide
excipient interaction and there is not enough
products. Further, in vivo excipient effects on
evidence to rule out the possibility of such an
permeability of furosemide cannot fully be ruled
interaction in vivo. It is noted that in vitro dissolution
out. Therefore, a biowaiver for the approval of
testing is not indicative for in vivo permeability
new multisource IR solid oral products containing
furosemide is inappropriate and BE should be
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 6, JUNE 2010
established with an in vivo BE study. This conclusion
man JB, Barends DM. 2009. Biowaiver monographs for
supports current regulatory guidances26–29 which do
immediate release solid oral dosage forms: Diclofenac sodium
not allow biowaivers for new multisource drug
and diclofenac potassium. J Pharm Sci 98:1206–1219.
9. Jantratid E, Strauch S, Becker C, Dressman JB, Amidon GL,
products containing BCS Class IV APIs.
Junginger HE, Kopp S, Midha KK, Shah VP, Stavchansky S,
Changes in approved drug products, such a change
Barends DM. 2009. Biowaiver monographs for immediate
in the manufacturing formula, in the manufacturing
release solid oral dosage forms: Doxycycline hyclate. J Pharm
process, in manufacturing sites and/or equipment
also necessitate demonstration of BE. If small, such
10. Becker C, Dressman JB, Amidon GL, Junginger HE, Kopp S,
Midha KK, Shah VP, Stavchansky S, Barends DM. 2008.
changes may be approvable without an in vivo BE
Biowaiver monographs for immediate release solid oral dosage
study. The FDA describes such postapproval changes
forms: Ethambutol dihydrochloride. J Pharm Sci 97:1350–
as SUPAC level 1 and level 2.100 The EU has a
comparable system.101 When a change to an approved
11. Potthast H, Dressman JB, Junginger HE, Midha KK, Oeser H,
furosemide IR drug product falls into such category,
Shah VP, Vogelpoel H, Barends DM. 2005. Biowaiver mono-graphs for immediate release solid oral dosage forms: Ibupro-
the data presented in this monograph (including the
excipient table for products with an MA) can be
12. Becker C, Dressman JB, Amidon GL, Junginger HE, Kopp S,
helpful to assess how critical the change is to product
Midha KK, Shah VP, Stavchansky S, Barends DM. 2007.
Biowaiver monographs for immediate release solid oral dosageforms: Isoniazid. J Pharm Sci 96:522–531.
13. Stosik AG, Junginger HE, Kopp S, Midha KK, Shah VP,
Stavchansky S, Dressman JB, Barends DM. 2008. Biowaiver
monographs for immediate release solid oral dosage forms:Metoclopramide hydrochloride. J Pharm Sci 97:3700–3708.
Kik Groot, RIVM, is acknowledged for producing
14. Vogt M, Derendorf H, Kra¨mer J, Junginger HE, Midha KK,
Shah VP, Stavchansky S, Dressman JB, Barends DM. 2007.
Biowaiver monographs for immediate release solid oral dosageforms: Prednisolone. J Pharm Sci 96:27–37.
15. Vogt M, Derendorf H, Kra¨mer J, Junginger HE, Midha KK,
Shah VP, Stavchansky S, Dressman JB, Barends DM. 2007. Biowaiver monographs for immediate release solid oral dosage
1. Vogelpoel H, Welink J, Amidon GL, Junginger HE, Midha KK,
forms: Prednisone. J Pharm Sci 96:1480–1489.
Mo¨ller H, Olling M, Shah VP, Barends DM. 2004. Biowaiver
16. Becker C, Dressman JB, Junginger HE, Kopp S, Shah VP,
monographs for immediate release solid oral dosage forms
Stavchansky S, Barends DM. 2008. Biowaiver monographs for
based on Biopharmaceutics Classification System (BCS). Lit-
immediate release solid oral dosage forms: Pyrazinamide.
erature data: Verapamil Hydrochloride, Propranolol Hydro-
chloride, and Atenolol. J Pharm Sci 93:1945–1956.
17. Grube S, Langguth P, Junginger HE, Kopp S, Midha KK,
2. Kalantzi L, Reppas C, Dressman JB, Amidon GL, Junginger
Shah VP, Stavchansky S, Dressman JB, Barends DM. 2009.
HE, Midha KK, Shah VP, Stavchansky SA, Barends DM.
Biowaiver monographs for immediate release solid oral dosage
2006. Biowaiver monographs for immediate release solid oral
forms: Quinidine sulfate. J Pharm Sci 98:2238–2251.
dosage forms: Acetaminophen (Paracetamol). J Pharm Sci
18. Korteja¨rvi H, Yliperttula M, Dressman JB, Junginger HE,
Midha KK, Shah VP, Barends DM. 2005. Biowaiver mono-
3. Granero GE, Longhi MR, Becker C, Junginger HE, Kopp S,
graphs for immediate release solid oral dosage forms: Raniti-
Midha KK, Shah VP, Stavchansky S, Dressman JB, Barends
dine hydrochloride. J Pharm Sci 94:1617–1625.
DM. 2008. Biowaiver monographs for immediate release solid
19. Becker C, Dressman JB, Junginger HE, Kopp S, Midha KK,
oral dosage forms: Acetazolamide. J Pharm Sci 97:3691–3699.
Shah VP, Stavchansky S, Barends DM. 2009. Biowaiver
4. Arnal J, Gonzalez-Alvarez I, Bermejo M, Amidon GL, Jungin-
monographs for immediate release solid oral dosage forms:
ger HE, Kopp S, Midha KK, Shah VP, Stavchansky S, Dress-
Rifampicine. J Pharm Sci 98:2252–2267.
man JB, Barends DM. 2008. Biowaiver monographs for
20. Sweetman S, editor. 2009. Martindale: The complete
drug reference. Electronic version. Pharmaceutical Press,
Thomson/MICROMEDEX, London, UK/Greenwood Village,
5. Manzo RH, Olivera ME, Amidon GL, Shah VP, Dressman JB,
Barends DM. 2006. Biowaiver monographs for immediate
21. Shin S-C, Kim J. 2003. Physicochemical characterization of
release solid oral dosage forms: Amitriptyline Hydrochloride.
solid dispersion of furosemide with TPGS. Int J Pharm
6. Verbeeck RK, Junginger HE, Midha KK, Shah VP, Barends
22. Rowbotham PC, Stanford JB, Sugden JK. 1976. Some aspects
DM. 2005. Biowaiver monographs for immediate release solid
of the photochemical degradation of frusemide. Pharm Acta
oral dosage forms based on biopharmaceutics classification
system (BCS) literature data: Chloroquine phosphate, chlor-
23. Devarakonda B, Otto DP, Judefeind A, Hill RA, de Villiers M.
oquine sulfate, and chloroquine hydrochloride. J Pharm Sci
2007. Effect of pH on the solubility and release of furosemide
from polyamidoamine (PAMAM) dendrimer complexes. Int J
7. Jantratid E, Prakongpan S, Dressman JB, Amidon GL,
Junginger HE, Midha KK, Barends DM. 2006. Biowaiver
24. Maestrelli F, Garcia-Fuentes M, Mura P, Alonso MJ. 2006.
monographs for immediate release solid oral dosage forms:
A new nanocarrier consisting of chitosan and hydroxypropyl-
Cimetidine. J Pharm Sci 95:974–984.
cyclodextrin. Eur J Pharm Biopharm 63:79–86.
8. Chuasuwan B, Binjesoh V, Polli JE, Zhang H, Amidon GL,
25. Motz SA. 2007. Combined assessment of dissolution and
Junginger HE, Midha KK, Shah VP, Stavchansky S, Dress-
epithelial permeability of solid oral dosage forms. Ph.D Thesis
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 6, JUNE 2010
Universita¨t Saarland. http://deposit.ddb.de/cgi-bin/dokserv?-
˜ ola de Medicamentos y Productos Sanitarios.
idn¼983385645&dok_var¼d1&dok_ext¼pdf&filename¼
http://www.agemed.es. (accessed May 15, 2009).
&filename¼ 983385645. pdf (accessed May 18, 2009).
46. La¨kemedelsverket. http://www.lakemedelsverket.se. (accessed
26. WHO. 2006. Multisource (generic) pharmaceutical products:
Guidelines on registration requirements to establish inter-
47. Datapharm Communications Ltd. http://www.medicines.org.
changeability. Technical Report Series, No. 937, 40th Report,
Annex 7 of WHO Expert committee on specifications for
48. DailyMed. http://www.dailymed.nlm.nih.gov. (accessed May
pharmaceutical preparations. http://whqlibdoc.who.int/trs/
WHO_TRS_937_eng.pdf. (accessed May 18, 2009).
49. Ponto LL, Schoenwald RD. 1990. Furosemide (frusemide). A
27. FDA. 2000. Guidance for Industry: Waiver of in vivo bioavail-
pharmacokinetic/pharmacodynamic review (Part I). Clin
ability and bioequivalence studies for immediate-release solid
oral dosage forms based on a Biopharmaceutics Classification
50. Ponto LL, Schoenwald RD. 1990. Furosemide (frusemide). A
System. US Food and Drug Administration, Center for Drug
pharmacokinetic/pharmacodynamic review (Part II). Clin
Evaluation and Research, USA. http://www.fda.gov/cder/
guidance/3618fnl.pdf. (accessed May 18, 2009).
51. Hammarlund MM, Paalzow LK, Odlind B. 1984. Pharmaco-
28. EMEA. 2001. Note for guidance on the investigation of bioa-
kinetics of furosemide in man after intravenous and oral
vailability and bioequivalence. http://www.emea.eu.int/pdfs/
administration: Application of moment analysis. Eur J Clin
29. EMEA. 2008. Draft Guideline on the Investigation of
52. Kelly MR, Cutler RE, Forrey AW, Kimpel BM. 1973. Phar-
Bioequivalence. CPMP/EWP/QWP/1401/98 Rev.1. (accessed
macokinetics of orally administered furosemide. Clin Phar-
May 18, 2009). http://www.emea.europa.eu/pdfs/human/qwp/
53. Grahne´n A, Hammarlund M, Lundqvist T. 1984. Implications
30. The United States Pharmacopeial Convention, Inc. 2009.
of intraindividual variability in bioavailability studies of fur-
USP;1; 32—NF 27. The United States Pharmacopeia—The
osemide. Eur J Clin Pharmacol 27:595–602.
National Formulary, Rockville MD 2085.
54. Chungi VS, Dittert LW, Smith RB. 1979. Gastrointestinal
31. Matsuda Y, Tatsumi E. 1990. Physicochemical characteriza-
sites of furosemide absorption in rats. Int J Pharm 4:27–38.
tion of furosemide modifications. Int J Pharm 60:11–
55. Clear NJ, Milton A, Humphrey M, Henry BT, Wulff M, Nichols
DJ, Anziano RJ, Wilding I. 2001. Evaluation of the intelisite
32. Beyers H, Malan SF, van der Watt JG, de Villiers MM. 2000.
capsule to deliver theophylline and frusemide tablets to the
Structure-solubility relationship and thermal decomposition
small intestine and colon. Eur J Pharm Sci 13:375–384.
of furosemide. Drug Dev Ind Pharm 26:1077–1083.
56. Waller ES, Crismon M, Smith R, Bauga M, Doluisio J. 1988.
33. Pudipeddi M, Serajuddin TM. 2005. Trends in solubility of
Comparative bioavailability of furosemide from solution and
polymorphs. J Pharm Sci 94:929–939.
40 mg tablets with different dissolution characteristics follow-
34. Latosinska JN, Latosinska M, Medycki W, Osuchowicz J.
ing oral administration in normal men. Biopharm Drug Dis
2006. Molecular dynamics of solid furosemide (4-chloro-2-
furfurylamino-5-sulfamoyl-benzoic acid) studied by NMR
57. McNamara PJ, Foster TS, Digenis GA, Patel RB, Craig WA,
and DFT methods. Chem Phy Lett 430:127–132.
Welling PG, Rapaka RS, Prasad VK, Shah VP. 1987. Influence
35. Berthod A. 1999. Hydrophobicity of ionizable compounds. A
of tablet dissolution of furosemide bioavailability: A bioequi-
theoretical study and measurements of diuretic octanol-water
valence study. Pharm Res 4:150–153.
partition coefficients by countercurrent chromatography.
58. Kelly MR, Cutler RE, Forrey AW, Kimpel BM. 1973. Phar-
macokinetics of orally administered furosemide. Clin Phar-
36. Ruiz-Angel MJ, Carda-Broch SC, Garcı´a-Alvarez-coque MC,
Berthod A. 2004. Micellar versus hydro-organic mobile phases
59. Cutler RE, Blair AD. 1979. Clinical pharmacokinetics of fru-
semide. Clin Pharmacokinet 4:279–296.
ionizable diuretics and an anionic surfactant. J Chromatogr
60. Beermann B, Midskov C. 1986. Reduced bioavailability and
effect of furosemide given with foof. Eur J Clin Pharmacol
37. Kasim NA, Whitehouse M, Ramachandran C, Bermejo M,
Lennerna¨s H, Hussain AS, Junginger HE, Stavchansky SA,
61. Waller ES, Massarella JW, Tomkiw MS, Smith RV, Doluisio
Midha KK, Shah VP, Amidon GL. 2004. Molecular properties
JT. 1985. Pharmacokinetics of furosemide after three different
of WHO essential drugs and provisional biopharmaceutical
single oral doses. Biopharm Drug Dispos 6:109–117.
classification. Mol Pharm 1:85–96.
62. Smith DE, Lin ET, Benet LZ. 1980. Absorption and disposition
38. WHO. WHO model list of essential medicines. 16th list, March
of furosemide in healthy volunteers, measured with a meta-
2009. http://www.who.int/selection_medicines/committees/
bolite-specific assay. Drug Metab Dis 8:337–342.
expert/17/WEB_unedited_16th_LIST.pdf. (accessed May 8,
63. Waller E, Hamilton SF, Massarella JW, Sharanevych MA,
Smith RV, Yakatan GJ, Doluisio JT. 1982. Disposition and
39. ROTE LISTE1 Arzneimittelverzeichnis fu¨r Deutschland.
absolute bioavailability of furosemide in healthy males.
http://www.rote-liste.de. (accessed May 8, 2009).
64. Lennernas H, Knutson L, Knutson T, Lesko L, Salmonson T,
Amidon GL. 1995. Human effective permeability data for
41. National Agency for Medicines. http://www.nam.fi. (accessed
furosemide, hydrochlothiazide, ketoprofen and naproxen to
be used in the proposed biopharmaceutical classification for IR
42. VIDAL. Fiches me´dicaments. http://www.vidal.fr. (accessed
65. Yamashita S, Furubayashi T, Kataoka M, Sakane T, Sezaki H,
43. College ter Beoordeling van Geneesmiddelen—Medicines
Tokuda H. 2000. Optimized conditions for prediction of intest-
Evaluation Board. http://www.cbg-meb.nl. (accessed May 8,
inal drug permeability using Caco-2 cells. Eur J Pharm Sci
44. Norwegian Medicines Agency. http://www.legemiddelverket.no.
66. Corti G, Maestrelli F, Cirri M, Zerrouk N, Mura P. 2006.
Development and evaluation of an in vitro method for
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 6, JUNE 2010
prediction of human drug absorption II. Demonstration on the
furosemide on some pharmacokinetic parameters. Int J Clin
method suitability. Eur J Pharm Sci 27:354–362.
67. Jung SJ, Choi SO, Um SY, Il Kim JI, Choo HYP, Cho SYi,
85. Rubinstein MH, Rughani JM. 1978. The effect of four tablet
Chung SY. 2006. Prediction of the permeability of drugs
binders on the bioavailability of frusemide from 40MG tablets.
through study on quantitative structure-permeability rela-
tionship. J Pharm Biom Anal 41:469–475.
86. Stu¨ber W, Mutschler E, Steinbach D. 1982. The pharmaceu-
68. Flanagan SD, Benet LZ. 1999. Net secretion of furosemide is
tical and biological availability of commercial preparations
subject to indomethacin inhibition, as observed in Caco-2
of furosemide. Arzneimittelforschung Drug Res 32:693–697.
monolayers and excised rat Jejunum. Pharm Res 16:221–224.
87. Prasad VK, Rapaka RS, Knight PW, Cabana BE. 1982. Dis-
69. Pade V, Stavchansky S. 1997. Estimation of the relative
solution medium—A critical parameter to identify bioavail-
contribution of the transcellular and paracellular pathway
ability problems of furosemide tablets. Int J Pharm 11:81–90.
to the transport of passively absorbed drugs in Caco-2 cell
88. Kingsford M, Eggers NJ, Soteros G. 1984. An in vivo–in vitro
culture model. Pharm Res 14:1210–1215.
correlation for the bioavailability of frusemide tablets.
70. Rege BD, Yu LX, Hussain AS, Polli JE. 2001. Effect of common
excipients on Caco-2 transport of low-permeability drugs.
89. Qureshi SA, McGilveray IJ. 1998. Assessment of pharmaceu-
tical quality of furosemide tablets from multinational mar-
71. Hilgendorf C, Spahn-Langguth H, Regardh CG, Lipka E,
kets. Drug Dev Ind Pharm 24:995–1005.
Amidon GL, Langguth P. 2000. Caco-2 versus Caco-2/HT29-
90. WHO. 2006. Proposal to waive in vivo bioequivalence require-
MTX co-cultured cell lines: Permeabilities via diffusion,
ments for WHO Model List of Essential Medicines immediate-
inside- and outside directed carrier-mediated transport.
release, solid oral dosage forms. Technical Report Series,
No937, 40th Report, Annex 8 of WHO Expert committee on
72. Winiwarter S, Bonham NM, Ax F, Hallberg A, Lennerna¨s H,
specifications for pharmaceutical preparations. http://whqlib-
Karle´n A. 1998. Correlation of human jejunal permeability (in
doc.who.int/trs/WHO_TRS_937_eng.pdf. (accessed July 22,
vivo) of drugs with experimentally and theoretically derived
parameters. A multivariate data analysis approach. J Med
91. Hammarlund-Idemaes M, Benet LZ. 1989. Furosemide phar-
macokinetics and pharmacodynamics in health and disease—
73. Collnot EM, Baldes C, Wempe MF, Hyatt J, Navarro L, Edgar
An update. J Biopharmacokinet Biopharm 17:1–46.
KJ, Friedrich Schaeffer U, Lehr CM. 2006. Influence of vita-
92. Artursson P, Palm K, Luthaman K. 2001. Caco-2 monolayers
min ETPGS poly(ethylene glycol) chain length on apical efflux
in experimental and theoretical predictions of drug transport.
transporters in Caco-2 cell monolayers. J Control Release
93. Rinaki E, Valsami G, Macheras P. 2003. Quantitative bio-
74. Beermann B, Dalen E, Lindstrom B. 1978. Bioavailability of
pharmaceutics classification system: The central role of dose/
two furosemide preparations. Br J Clin Pharmacol 6:537–538.
solubility ratio. Pharm Res 20:1917–1925.
75. Eggers NJ, Kingsford M, Saint Joly CM, Jellett LB, Maling
94. Gres M-C, Julian B, Bourre M, Meunier V, Roques C, Berger
TJ. 1980. Bioavailability of furosemide. N Z Med J 91:403–404.
M, Boulenc X, Berger Y, Fabre G. 1998. Correlation between
76. Habermann W, Rudolph F. 1980. Bioequivalence studies with
oral drug absorption in humans and apparent drug perme-
ability in TC-7 cells, a human epithelial intestinal cell line:
77. Martin BK, Uihlein M, Ings RM, Stevens LA, McEwen J. 1984.
Comparison with the parental Caco-2 cell line. Pharm Res
Comparative bioavailability of two furosemide formulations in
95. Lindenberg M, Kopp S, Dressman JB. 2004. Classification of
78. Straughn AB, Wood GC, Raghow G, Meyer MC. 1986. Bioa-
orally administered drugs on the World Health Organization
vailability of seven furosemide tablets in man. Biopharm Drug
model list of essential medicines according to the biopharma-
ceutics classification system. Eur J Pham Biopharm 58:265–
79. Kaojarern S, Poobrasert O, Utiswannakul A, Kositchaiwat U.
1990. Bioavailability and pharmacokinetics of furosemide
96. Wu C-Y, Benet LZ. 2005. Predicting drug disposition via
marketed in Thailand. J Med Assoc Thai 73:191–197.
application of BCS: Transport/absorption/elimination inter-
80. Awad R, Arafat T, Saket M, Saleh M, Gharaibeh M, Zmeili S,
play and development of a biopharmaceutics drug disposition
Sallam E, Shubair M, Qobrosi S. 1992. A bioequivalence study
classification system. Pharm Res 22:11–23.
of two products of furosemide tablets. Int J Clin Pharmacol
97. Schulz M, Schmoldt A. 2003. Therapeutic and toxic blood
concentrations of more than 800 drugs and other xenobiotics.
81. Nakib N, Idkaidek N, Beshtawi M, Bader M, Admour I, Alam
SM, Zaman Q, Dham R. 2003. Bioequivalence evaluation of
98. FDA. Code of Federal Regulations. Title 21, Part 320: Bio-
two brands of furosemide 40 mg tablets (Salurin and Lasix)
availability and Bioequivalence requirements. Section 320.33.
in healthy human volunteers. Biopharm Drug Dis 24:245–
2003. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/
CFRSearch.cfm?fr¼320.33. (accessed May 18, 2009).
82. Cuadrado A, Rodrı´guez Gasco´n A, Herna´ndez RM, Castilla
99. PAHO. Science based criteria for bioequivalence in vivo and in
AM, de la Maza A, Ya´nez C, Lo´pez de Oca´riz A, Solinı´s MA,
vitro, biowaivers, and strategic framework for implementa-
Pedraz JL. 2003. In vivo pharmacokinetic-pharmacodynamic
http://www.paho.org/english/ad/ths/ev/be-doct-draft-
relationship and in vitro equivalence of two oral furosemide
tablet formulations. Arzneimittelforschung Drug Res 53:321–
100. FDA. 2005. Guidance for Industry. Immediate Release Solid
Oral Dosage Forms Scale-Up and Postapproval Changes:
83. Yu LX. 2004. BioINequivalence: Concept and Definition.
Chemistry, Manufacturing, and Controls, In Vitro Dissolution
ACPS Meeting, October 19–20. http://www.fda.gov/ohrms/
(SUPAC-IR). www.fda.gov/cder/guidance/supac.htm.
101. European Commission. 2006. Guideline on Dossier require-
84. Wolf-Coporda A, Lovric´ Z, Huic´ M, Francetic´ I, Vrhovac B,
ments for Type IA and Type IB. http://ec.europa.eu/enterprise/
Plavsic´ F, Skreblin M. 1996. Determination of bioequivalence
pharmaceuticals/eudralex/vol-2/c/var_type_1a1b_guideline_06-
of two furosemide preparations; the effect of high doses of
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 99, NO. 6, JUNE 2010
PETERS, F.E. Os monoteístas, volume 1: os povosde Deus. São Paulo, Editora Contexto, 2007, 384páginas, ISBN 97885722443654. R$ 59,00. O tema do monoteísmo fascina, intriga e inspira. Crentes ou não, a fé em um único Deus apresenta facetasque atraíram os estudiosos e as pessoas comuns. A crençaem muitos poderes sobrenaturais foi predominante, emtantos povos, por tantos séculos. Deuses e
Extract from Course F18: Furnace brazing stainless steels for automotive applications ………………………There are two atmosphere categories that have to be considered. These are: 1. Chemically active atmospheres. 2 . Chemically inert atmospheres (including vacuum!). 1. Chemically active atmospheres Atmospheres of this type react with the oxides
 evaluation of these documents. Biowaiver mono-
headache, hypotension, muscle cramps, dry mouth,
graphs have already been published for acetamino-
thirst, weakness, etc.20 There is generally no need to
phen (INN: paracetamol),2 acetazolamide,3 aciclovir,4
amitriptyline,5 atenolol,1 chloroquine,6 cimetidine,7diclofenac,8 doxycyline hyclate,9 ethambutol,10 ibu-
profen,11 isoniazid,12 metoclopramide,13 predniso-lone,14 prednisone,15 pyrazinamide,16 propranolol,1
quinidine,17 ranitidine,18 rifampicin,19 and verapa-mil.1 They are also available on-line at www.fip.
evaluation of these documents. Biowaiver mono-
headache, hypotension, muscle cramps, dry mouth,
graphs have already been published for acetamino-
thirst, weakness, etc.20 There is generally no need to
phen (INN: paracetamol),2 acetazolamide,3 aciclovir,4
amitriptyline,5 atenolol,1 chloroquine,6 cimetidine,7diclofenac,8 doxycyline hyclate,9 ethambutol,10 ibu-
profen,11 isoniazid,12 metoclopramide,13 predniso-lone,14 prednisone,15 pyrazinamide,16 propranolol,1
quinidine,17 ranitidine,18 rifampicin,19 and verapa-mil.1 They are also available on-line at www.fip.