0022-3565/98/2863-1146$03.00/0THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS
Copyright 1998 by The American Society for Pharmacology and Experimental Therapeutics
Evidence against Anandamide as the Hyperpolarizing FactorMediating the Nitric Oxide-Independent Coronary VasodilatorEffect of Bradykinin in the Rat1
Department of Pharmacology and Molecular Cardiobiology Division, Boyer Center for Molecular Medicine, Yale University, New Haven,Connecticut (D.F.) and Department of Cell Biology, UMDNJ-SOM, Stratford, New Jersey (J.Q.)
This paper is available online at http://www.jpet.org
ABSTRACT The mediator of nitric oxide-(NO) independent vasodilation at-
nist, SR 141716A (2 M), reduced dose-dependent vasodilator
tributed to endothelium-derived hyperpolarizing factor remains
responses to anandamide (1–10 g) but was without effect on
unidentified although there is evidence for a cytochrome P450-
responses to AA (1–10 g), bradykinin (10 –1000 ng) or cro-
derived eicosanoid. Anandamide, the ethanolamide of arachi-
makalim (1–10 g). Inhibition of voltage-dependent Caϩϩ
donic acid and an endogenous ligand for cannabinoid recep-
channels with nifedipine (5 nM) attenuated vasodilation to
tors, was proposed as an endothelium-derived hyperpolarizing
anandamide and arachidonic acid whereas inhibition of Caϩϩ-
factor-mediating mesenteric vasodilation to acetylcholine and
activated Kϩ channels with charybdotoxin (10 nM) reduced
the hypotensive effect of bradykinin. Using pharmacological
responses to arachidonic acid but had no effect on vasodilation
interventions that attenuate responses to bradykinin, we exam-
induced by anandamide. Inhibition of cytochrome P450 with
ined the possibility of anandamide as a mediator of the NO-
clotrimazole (1 M) greatly reduced vasodilator responses to
independent vasodilator effect of bradykinin in the rat perfused
bradykinin with less effect on those to anandamide. Finally, the
heart by determining responses to anandamide and arachi-
time course of the coronary vasodilator responses to anand-
donic acid. Hearts were treated with indomethacin to exclude
amide and bradykinin were dissimilar. These results argue
prostaglandins and nitroarginine to inhibit NO synthesis and
against a role of anandamide in the vasodilator effect of bra-
elevate perfusion pressure. The cannabinoid receptor antago-
The recognition by Furchgott and Zawadski (1980) of the
hypothesis (Zygmunt et al., 1996; Edwards et al., 1996;
requirement for an intact endothelium for responses to cer-
Fukao et al., 1997). Our studies with bradykinin in the rat
tain vasodilator agonists led to the identification of NO as
heart and/or kidney demonstrate that the NO-independent
EDRF. The introduction of inhibitors of NO synthesis under-
vasodilator effect of this peptide is susceptible to inhibitors
scored the importance of NO to the regulation of vascular
PLC and PLA , P450 and Kϩ channels, supporting the con-
tone. However, their use also resulted in the realization that
cept of a P450-derived eicosanoid as a hyperpolarizing factor
NO could not fully account for endothelium-dependent re-
(Fulton et al., 1992, 1994, 1995, 1996; Rapacon et al., 1996).
sponses to various agonists including bradykinin and acetyl-
Studies using inhibitors of P450 that exhibit differential
choline, depending on the vascular bed and the species. Con-
activity against epoxygenase vs. w-hydroxylase, i.e., clotrim-
sequently, release of an unidentified hyperpolarizing factor,
azole vs. 17-ODYA (Fulton et al., 1995), suggest that of the
a term first coined by Taylor and Weston (1988), was in-
AA metabolites, an EET is the most likely candidate. More-
over, GC-MS analysis of coronary perfusates revealed the
Currently, there is considerable support for a P450-derived
release of EETs but not HETEs. EETs are vasodilator, syn-
metabolite of AA as an EDHF (Bauersachs et al., 1994;
thesized by the endothelium and stimulate Caϩϩ-activated
Hecker et al., 1994; Campbell et al., 1996; Popp et al., 1996)
Kϩ channels (Campbell et al., 1996). Our pharmacological
although problems with the specificity of inhibitors of P450
studies indicate that, of the four EET regioisomers, only 5,6
have culminated in several recent studies that question this
EET can fulfill the criteria for a putative mediator of thecoronary vasodilator effect of bradykinin (Quilley et al.,
Received for publication January 26, 1998. 1
This work was supported by National Institutes of Health Grant 49275
and American Heart Association Grant 940-318.
However, there are major reservations concerning the pro-
ABBREVIATIONS: EDHF, endothelium-derived hyperpolarizing factor; AA, arachidonic acid; NO, nitric oxide; P450, cytochrome P450; EET, epoxide; HETE, hydroxyeicosatetraenoic acid; PLC; phospholipase C; PLA , phospholipase A ; GC-MS, gas chromatography-mass spectrometry. Anandamide and Vasodilation to BK
posal that a P450-AA metabolite may be a hyperpolarizing
n ϭ 4), charydotoxin (10 nM; n ϭ 5) and SR 141716A (2 M; n ϭ 6).
factor. These revolve primarily around the limited specificity
Thus, we have previously reported that the coronary vasodilator
of the inhibitors that have been used to implicate P450 (Ed-
activity of bradykinin is reduced by nifedipine and charybdotoxin
wards et al., 1996; Fukao et al., 1997; Ohlmann et al., 1997).
(Fulton et al., 1994) whereas the vasodilator effect of anandamide in
As a result, other potential mediators have been sought and
the rat mesenteric vascular bed is inhibited by SR 141716A (Randallet al., 1996). The antagonists were added to the perfusate at least 10
Randall et al. (1996) proposed that anandamide, the ethano-
min before obtaining responses to anandamide and AA. The concen-
lamide of AA and the putative endogenous ligand for canna-
tration of SR 141716A was twice that used by Randall et al. (1996)
binoid receptors (Devane et al., 1992), may be an EDHF in
whereas the concentration of nifedipine and charybdotoxin were
the rat. Thus, a product with the chromatographic properties
those we had previously shown to inhibit coronary vasodilator re-
of authentic anandamide was released from the perfused
sponses to bradykinin (Fulton et al., 1994). Three to four prepara-
mesenteric vascular bed labeled with 3H-AA and challenged
tions per day were completed and at least one served as a control; the
with carbachol and the mesenteric vasodilator effect of anan-
others were assigned randomly to each of the treatment groups. In
damide was greatly reduced in the presence of depolarizing
the experiments with SR 141716A, responses to nitroprusside (1 g)
concentrations of KCl, suggesting a role for activation of Kϩ
were used an index of effects apparently unrelated to antagonism of
channels (Randall et al., 1996). This group also reported that
cannabinoid receptors. In the experiments with nifedipine and SR141716A which both reduced coronary vascular tone, U46619 was
the NO-independent hypotensive effect of bradykinin in the
added to the perfusate (10 ng/ml for nifedipine and 0.5–1.0 ng/ml for
anesthetized rat was attenuated by pretreatment with a
SR 141716A) to restore perfusion pressure to its previous level.
cannabinoid receptor antagonist that also blocked the effect
In a second series of experiments, we compared the effects of SR
of anandamide in the mesenteric vasculature (Randall et al.,
141716A (2 M; n ϭ 4) or vehicle (n ϭ 4) on coronary vasodilator
1996). In contrast to these studies in the rat, Pratt et al.
responses to bradykinin (10 –1000 ng) as the hypotensive response to
(1998) reported that the vasorelaxant effect of anandamide in
bradykinin in anesthetized rats has been reported to be attenuated
the bovine coronary artery was independent of cannabinoid
by pretreatment with SR 141517A (Randall et al., 1996). Responses
receptors but involved the release of AA and its subsequent
to cromakalim (1, 3 and 10 g) were used to assess any direct effects
conversion to vasodilatory eicosanoids.
of SR 141716A on Kϩ channels and unrelated to cannabinoid recep-
Consequently, we used pharmacological criteria, based on
In a third series of experiments, vasodilator responses to anand-
our studies with bradykinin, to examine whether anandam-
amide (3 and 10 g) and bradykinin (30 and 100 ng) were compared
ide could fulfill the requirements for a putative mediator for
in the absence (n ϭ 6) and presence (n ϭ 5) of the P450 inhibitor,
bradykinin-induced vasodilation in the isolated heart of the
clotrimazole (1 M), as coronary vasodilator responses to bradykinin
rat. Thus, we determined coronary vasodilator responses to
have been shown to be attenuated by clotrimazole (Fulton et al.,
anandamide in the presence and absence of nifedipine to
1995) and anandamide has been reported to be a substrate for P450
prevent vasodilation resulting from closure of voltage-depen-
(Bornheim et al., 1993). However, if anandamide is the mediator of
dent Caϩϩ channels, charydotoxin to inhibit Caϩϩ-activated
bradykinin-induced vasodilation, then clotrimazole should be with-
Kϩ channels and SR 141716A to antagonize cannabinoid
out effect on responses to anandamide although attenuating those to
receptors. The effects of SR 141517A on responses to brady-
bradykinin. We chose clotrimazole, despite reports of effects on Kϩ
kinin were also determined. As anandamide is readily
channels, because it is considered to be more specific for epoxygenasethan -hydroxylase. Moreover, at the concentration chosen (1 M),
cleaved by an amidase to yield AA, the effects of these inter-
we have no evidence for effects on Kϩ channels as clotrimazole did
ventions on responses to AA were also examined. We also
not affect vasodilator response to cromakalim or SCA 40 (Fulton et
compared the effects of a P450 inhibitor, clotrimazole, on
al., 1994) which has been reported to stimulate Caϩϩ-activated Kϩ
responses to bradykinin and anandamide as this compound
channels (Laurent et al., 1993).
is a substrate for P450 (Bornheim et al., 1993). The results
Statistics. Vasodilator responses in control and treatment groups
indicate that anandamide is unlikely to be the mediator of
were compared by analysis of variance and individual points were
bradykinin-induced, NO-independent vasodilation in the rat
compared by Neuman-Keuls test. Differences were considered sta-
tistically significant when P Ͻ .05. Materials. Anandamide was obtained from Biomol (Plymouth
Meeting, PA) and was dissolved in ethanol. Indomethacin, nitroargi-
nine, bradykinin, nifedipine, cromakalim, clotrimazole and nitro-prusside were purchased from Sigma Chemical Co. (St. Louis, MO).
Male Wistar rats, weight 360 to 460 g, were anaesthetized with
Indomethacin was dissolved in 4.2% NaHCO , clotrimazole in etha-
pentobarbital, 65 mg/kg i.p., and heparin, 1000 U/kg, was adminis-
nol and cromakalim in ethanol before dilution with saline. The other
tered i.v. After thoracotomy, the heart with attached aorta was
agents were dissolved in distilled water. Charybdotoxin was pur-
excised and flushed free of blood with ice-cold Krebs’ buffer. The
chased from Peptides International (Louisville, KY) and was dis-
heart was then cannulated via the aorta and perfused retrogradely
solved in distilled water. SR141716A was a gift from RBI (Natick,
with oxygenated Krebs’ buffer at 37°C at a constant flow rate (8 –10
MA) supported by NIMH Chemical Synthesis Program and was
ml/min) to obtain an initial basal perfusion pressure of 30 to 40
dissolved in ethanol. U46619 was obtained from UpJohn (Kalmazoo,
mmHg. The perfusate contained indomethacin (2.8 M) to inhibit
MI) and was dissolved in ethanol and diluted with distilled water.
cyclooxygenase and nitroarginine (50 M) was added to inhibit NO
Arachidonic acid (NuChek, Elysian, MN) was dissolved in distilled
synthase and elevate perfusion pressure to 130 to 140 mmHg and
also to reproduce the experimental conditions that were used toaddress the mechanism of bradykinin-induced vasodilation (Fultonet al., 1994, 1995, 1996).
Once a stable elevated perfusion pressure was obtained, vasodila-
tor responses to increasing doses of anandamide (1, 3 and 10 g)
Initial basal perfusion pressures were not different in the
were determined followed by responses to increasing doses of AA (1,
various groups: vehicle, 37 Ϯ 2 mmHg; SR 141716A, 38 Ϯ 2
3 and 10 g) in the absence (n ϭ 8) and presence of nifedipine (5 nM;
mmHg; charybdotoxin, 39 Ϯ 2 mmHg and nifedipine, 42 Ϯ 3
Fulton and Quilley
mmHg. Elevated perfusion pressures were comparable in all
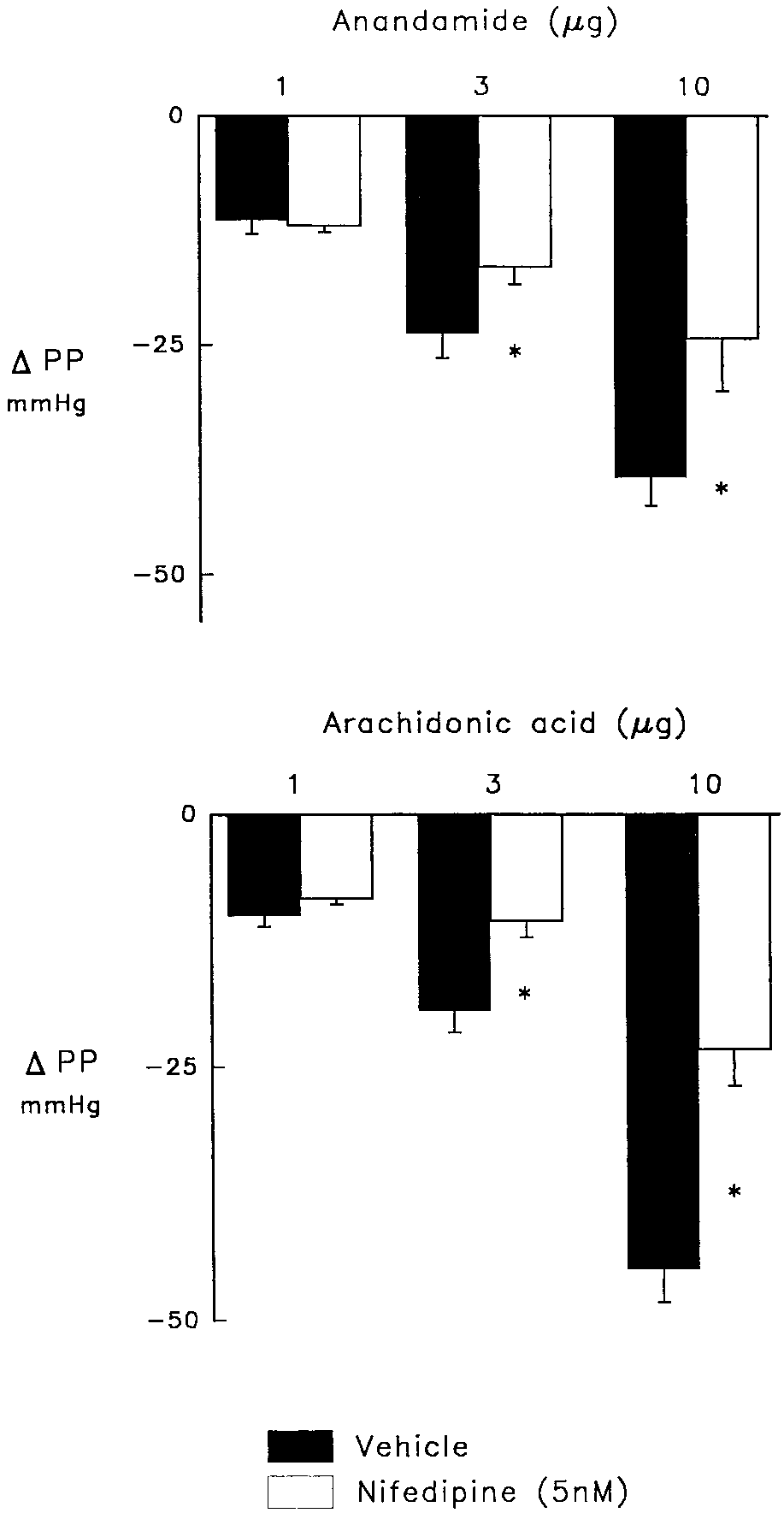
Inhibition of voltage-dependent Caϩϩ channels with nifed-
the groups except the charybdotoxin group where pressure
ipine diminished the vasodilator effects of 3 and 10 g anan-
was further increased by inhibition of Caϩϩ-activated Kϩ
damide (P Ͻ .05) and AA (P Ͻ .05) to a similar degree without
channels to 155 Ϯ 4 mmHg compared to 134 Ϯ 2 mmHg for
affecting the responses to the lowest doses of these agents
vehicle, 138 Ϯ 3 mmHg for SR 141716A and 131 Ϯ 6 mmHg
Inhibition of Caϩϩ-activated Kϩ channels with charydo-
In the vehicle control group, 1, 3 and 10 g anandamide
toxin did not reduce the coronary vasodilator response to
elicited dose-dependent falls in perfusion pressure of 11 Ϯ 2,
anandamide (fig. 3), rather, the response to the lowest dose of
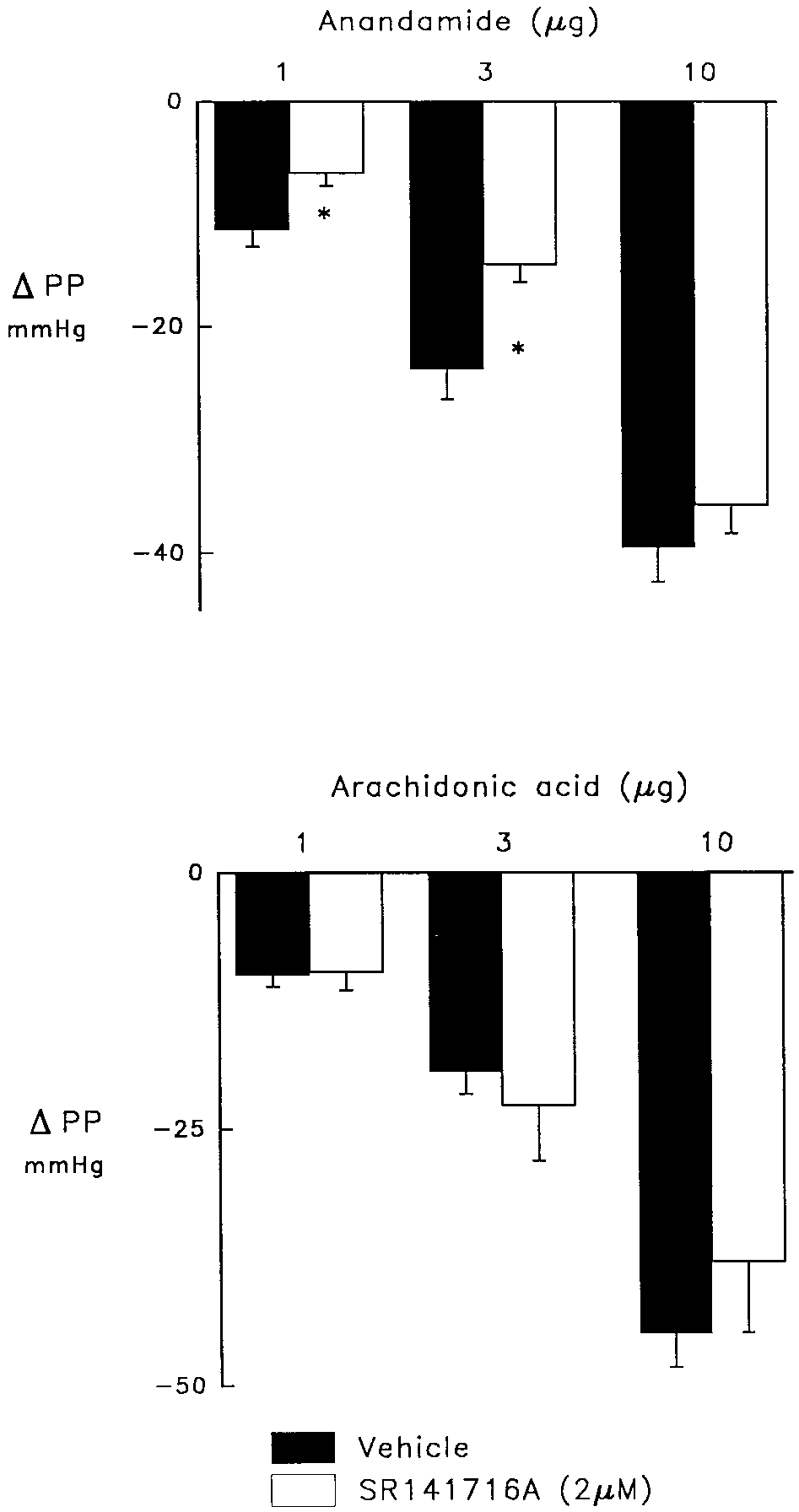
24 Ϯ 3 and 40 Ϯ 3 mmHg, respectively (fig. 1). The cannabi-
anandamide was slightly increased from 11 Ϯ 2 to 16 Ϯ 2
noid receptor antagonist, SR 141716A, reduced the coronary
mmHg (P Ͻ .05). In contrast, the coronary vasodilator effect
vasodilator response to the two lower doses of anandamide,
of AA was significantly reduced in the presence of charydo-
6 Ϯ 1 and 15 Ϯ 2 mmHg (P Ͻ .05), but was without effect on
the highest dose, 36 Ϯ 3 mmHg. In contrast, the dose-depen-
In the second series of experiments to determine the effects
dent coronary vasodilator response to AA was unaffected by
of SR 141716A on vasodilator responses to bradykinin and
SR 141716A (fig. 1). SR 141716A did not affect vasodilator
cromakalim, basal and elevated perfusion pressures in the
responses to nitroprusside, 37 Ϯ 4 vs. 44 Ϯ 7 mmHg for the
control and treatment groups were 34 Ϯ 2 and 134 Ϯ 6
Fig. 2. Vasodilator responses to anandamide (upper panel) and arachi- donic acid (lower panel) in control hearts (solid bars) and in the presence Fig. 1. Effect of the cannabinoid receptor antagonist, SR 141716A, (2 M;
of nifedipine (5 nM; open bars). Heart were treated with indomethacin
open bars) or vehicle (solid bars) on vasodilator responses to anandamide
(2.8 M) and nitroarginine (50 M) to inhibit cyclooxygenase and NO
(upper panel) and arachidonic acid (lower panel) in the rat isolated
synthase and elevate perfusion pressure from 30 – 40 to 130 –140 mmHg.
perfused heart treated with indomethacin (2.8 M) and nitroarginine (50
Nifedipine reduced elevated perfusion pressure that was restored with
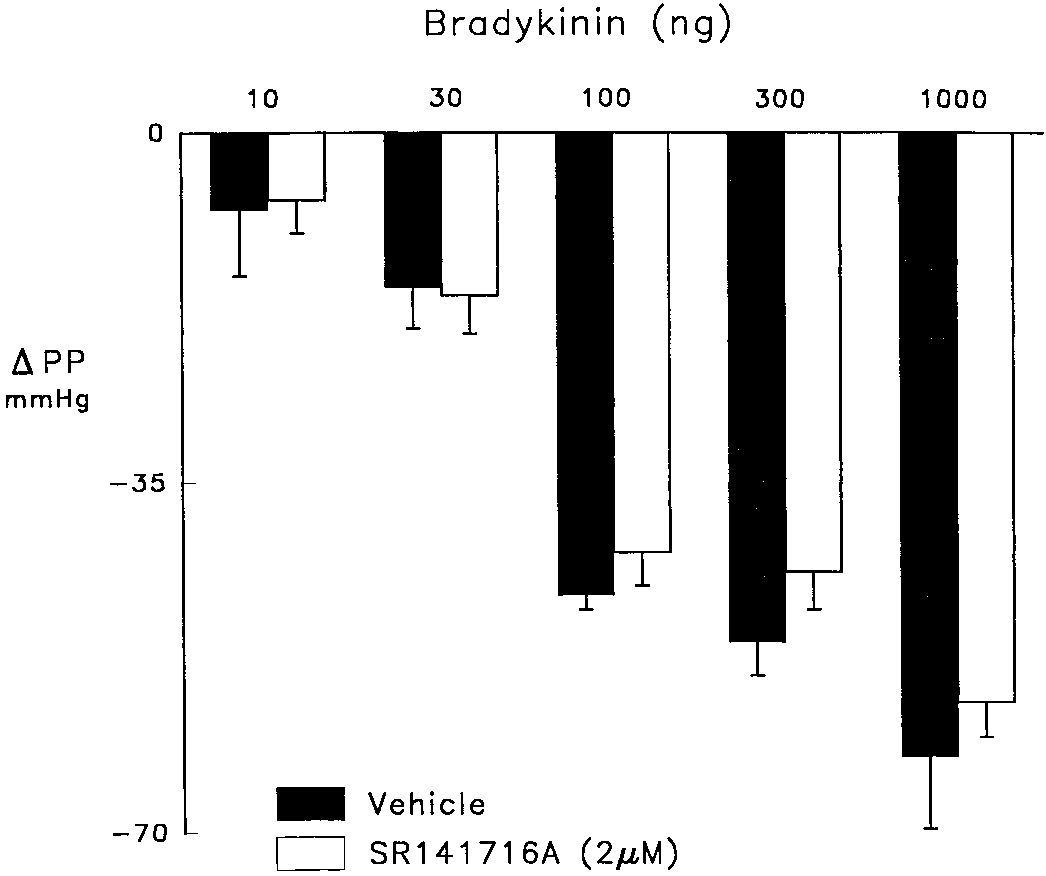
M) which elevated perfusion pressure from 30–40 to 130–140 mmHg. Anandamide and Vasodilation to BK Fig. 4. Vasodilator responses to bradykinin in control hearts (solid bars) and those treated with SR 141716A (2 M; open bars). The coronary perfusate contained indomethacin (2.8 M) and nitroarginine (50 M) to inhibit cyclooxygenase and NO synthase and elevate perfusion pressure from 30 – 40 to 130 –140 mmHg. SR 141716A reduced elevated perfusion pressure which was restored with U46619 (0.5–1.0 ng/ml).
sure in the control group was 131 Ϯ 1 mmHg compared to128 Ϯ 2 mmHg in the clotrimazole group.
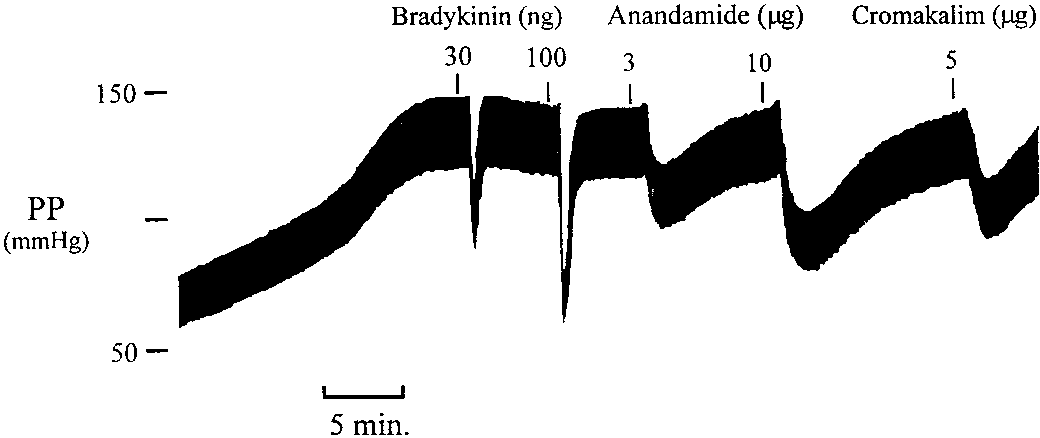
Figure 5 shows a recording of perfusion pressure from a
vehicle-treated heart and the vasodilator responses to brady-kinin and anandamide. The response to bradykinin wasrapid in onset and of short duration whereas the response toanandamide developed more slowly and was of longer dura-tion. Discussion
Several studies have provided evidence to support a P450-
derived metabolite of AA as an EDHF mediating the NO-independent vasodilator/vasorelaxant response to bradyki-
Fig. 3. Effects of charybdotoxin (10 nM; open bars) compared to vehicle (solid bars) on vasodilator responses to anandamide (upper panel) or
nin and/or acetylcholine (Hecker et al., 1994; Bauersachs et
arachidonic acid (lower panel) in the isolated perfused heart treated with
al., 1995; Campbell et al., 1996; Popp et al., 1996). Our
indomethacin (2.8 M) and nitroarginine (50 M) to inhibit cyclooxygen-
studies are consistent with this concept as the coronary
ase and NO synthase and elevate perfusion pressure from 30 – 40 to130 –140 mmHg. Charybdotoxin caused a further elevation of perfusion
and/or renal vasodilator action of bradykinin is susceptible to
inhibitors of PLC and PLA , P450 and Caϩϩ-activated Kϩ
channels (Fulton et al., 1992, 1994, 1995, 1996). Of the
mmHg, respectively, and 36 Ϯ 2 and 138 Ϯ 5 mmHg, respec-
P450-AA metabolites, an EET is considered the most likely
tively. SR 141716A did not affect responses to bradykinin
as an EDHF as EETs are produced by the endothelium and
(fig. 4) but tended to reduce those to cromakalim although
are vasodilator, presumably by their ability to activate Kϩ
the differences were not significant. In control hearts, 1, 3and 10 g cromakalim decreased perfusion pressure by 6 Ϯ 1,22 Ϯ 3 and 58 Ϯ 7 mmHg, respectively, compared to 5 Ϯ 3,13 Ϯ 4 and 43 Ϯ 4 mmHg, respectively, for hearts treatedwith SR 141716A.
In the presence of clotrimazole to inhibit P450, vasodilator
responses to bradykinin were almost abolished, confirmingour previous results (Fulton et al., 1995). Thus, reductions inperfusion pressure to 30 and 100 ng bradykinin were 2 Ϯ 1and 6 Ϯ 2 mmHg, respectively, compared to control values of20 Ϯ 3 and 39 Ϯ 4 mmHg, respectively. Clotrimazole alsoreduced coronary vasodilator responses to 3 and 10 g anan-
Fig. 5. Recording of perfusion pressure in response to bradykinin, anan- damide and cromakalim in the isolated heart treated with indomethacin
damide from 21 Ϯ 2 and 33 Ϯ 2 mmHg, respectively, to 12 Ϯ
(2.8 M) and nitroarginine (50 M) to inhibit prostaglandin and NO
1 and 23 Ϯ 1 mmHg, respectively. Elevated perfusion pres-
synthesis and elevate perfusion pressure to approximately 130 mmHg. Fulton and Quilley
channels (Hu and Kim, 1993; Campbell et al., 1996). How-
ization, for example. However, the Kϩ channels responsible
ever, a role of P450 has been questioned as inhibitors of this
for the effects of anandamide and AA must be different,
pathway exhibit a variety of actions apparently unrelated to
based on the results with charybdotoxin that markedly re-
inhibition of P450 and including effects on Kϩ channels (Oye-
duced the coronary vasodilator effect of AA but not that of
kan et al., 1994; Edwards et al., 1996). Moreover, the admin-
anandamide. These observations are good evidence against
istration of EETs has been reported to be without effect in
anandamide as a source of AA which then exerts a direct
some vascular preparations (Zygmunt et al., 1996). Conse-
effect or serves as a precursor for the formation of a product
quently, alternative mediators have been sought and Randall
that elicits vasodilation via a charybdotoxin-sensitive mech-
et al. (1996) proposed the ethanolamide of AA (anandamide)
anism. The alternative explanation, that AA stimulates an
which is the putative endogenous ligand for cannabinoid
endothelial Kϩ channel to initiate the release of a vasodilator
receptors. We considered anandamide an attractive possibil-
is untenable as inhibition of cannabinoid receptors with
ity for mediating vasodilator responses to bradykinin be-
SR141716A reduced responses to anandamide but failed to
cause our previous results would not be inconsistent with
influence responses to AA. The effect of SR141716A to reduce
this concept; as an analogue of AA, anandamide would pre-
responses to anandamide is unlikely to be due to an effect on
sumably be stored in phospholipids and released by the ac-
Kϩ channels as SR141716A did not affect responses to cro-
tions of phospholipases whereupon it could also serve as a
makalim and did not alter responses to bradykinin or AA
substrate for P450 (Bornheim et al., 1993) to produce a va-
which are dependent on activation of Kϩ channels.
sodilator that activates Kϩ channels.
Finally, we addressed the effect of an inhibitor of P450,
To address this possibility, we determined the vasodilator
clotrimazole, on the coronary vasodilator action of anandam-
activity of anandamide in the presence of pharmacological
ide as we have previously shown this agent reduces the
interventions that inhibit NO-independent coronary vasodi-
coronary and renal vasodilator actions of bradykinin. If
lator responses to bradykinin. Under identical experimental
anandamide itself is the mediator of the bradykinin effect,
conditions of inhibition of prostaglandin and NO synthesis,
then inhibition of P450 with clotrimazole should be without
coronary vasodilator responses to anandamide were tested
effect. Alternatively, if anandamide, after its release in re-
after treatment of hearts with nifedipine, charybdotoxin, clo-
sponse to bradykinin, requires conversion by P450 for activ-
trimazole and SR141716A and compared to those obtained
ity, then clotrimazole should inhibit the vasodilator effect of
with AA or bradykinin. The results obtained argue against
both anandamide and bradykinin to the same degree. Clo-
anandamide as the mediator of bradykinin-induced vasodi-
trimazole virtually abolished vasodilator responses to brady-
lation. First, inhibition of Caϩϩ-activated Kϩ channels with
kinin in this series of experiments, consistent with our pre-
charybdotoxin at a concentration that almost abolished cor-
vious observations (Fulton et al., 1995). In contrast,
onary vasodilator responses to bradykinin (Fulton et al.,
inhibition of vasodilation induced by anandamide was much
1994) was without effect on vasodilator responses to anand-
less pronounced, a result that provides further evidence
amide. The only explanation for these observations that per-
against anandamide as the mediator for bradykinin. How-
mits consideration of anandamide as the vasodilator media-
ever, the observation that clotrimazole reduced the vasodila-
tor activity of anandamide indicates that an intact P450
charydotoxin-sensitive Kϩ channel in the endothelium to
system may be required. Thus, anandamide can be a sub-
result in the release of the mediator, in this case anandam-
strate for P450 (Bornheim et al., 1993) although the activity
ide. In this scenario, administration of the mediator, anand-
of any products to elicit vasodilation remains to be deter-
amide, would by-pass the processes involved in its synthesis
mined. It is unlikely that anandamide first releases AA
and/or release. Consequently, any intervention that modifies
which is then converted by P450 to vasodilatory eicosanoids
the response to anandamide should also modify that to the
as suggested by Pratt et al. (1998) because charybdotoxin
initiating stimulus, i.e., bradykinin. However, the failure of
failed to affect dilator responses to anandamide but inhibited
the cannabinoid receptor antagonist, SR 141716A, to inhibit
those to AA. An alternative explanation for the inhibitory
the vasodilator effect of bradykinin although reducing that to
effects of clotrimazole on vasodilation induced by anandam-
anandamide argues against this possibility regardless of
ide is that clotrimazole exerts effects on Kϩ channels or even
whether the effect of SR 141716A is via inhibition of canna-
the cannabinoid receptor in addition to inhibiting P450.
binoid receptors or an alternative mechanism. Thus, the in-
The results from this study, therefore, do not support the
hibitory effect of SR 141716A on responses to anandamide
hypothesis proposed by Randall et al. (1996) that anandam-
was not pronounced and may reflect functional antagonism
ide is an EDHF in the rat. However, they used the perfused
(White and Hiley, 1997). Nonetheless, if anandamide is the
mesentery and studied vasodilation to acetylcholine which
mediator of bradykinin-induced vasodilation, then SR
was inhibited by SR141716A as was the endothelium-inde-
141716A should also attenuate the response to bradykinin
pendent vasodilator effect of anandamide, suggesting a role
for CB receptors. This is in contrast to our studies where the
The possibility that anandamide yields AA that then un-
effects of bradykinin in the heart were addressed and which
dergoes transformation by P450 to generate a vasodilator
provides a possible explanation for the different results.
product was also addressed in this study. Thus, the relatively
Thus, depending on the tissue and the agonist, different
slow onset of vasodilation to anandamide compared with
hyperpolarizing factors may be involved. However, the obser-
bradykinin is consistent with conversion to an active product.
vation of Randall et al. (1996) that the NO-independent hy-
The observation that nifedipine reduced the coronary vaso-
potensive effect of bradykinin in the rat is also attenuated by
dilator effects of both anandamide and AA is consistent with
SR141716A, suggesting a mechanism operating through can-
a common vasodilator mechanism that involves closure of
nabinoid receptors, is not supported by our study.
voltage-dependent Caϩϩ channels in response to hyperpolar-
In summary, our observations, when viewed collectively,
Anandamide and Vasodilation to BK
strongly suggest that anandamide is unlikely to be the EDHF
Fulton D, McGiff JC and Quilley J (1996) Role of phospholipase C and phospholipase
mediating the NO-independent vasodilator effect of bradyki-
A2 in the nitric oxide-independent vasodilator effect of bradykinin in the rat perfused heart. J Pharmacol Exp Ther 278:518 –526.
nin in the rat heart and support the conclusions reached by
Furchgott RF and Zawadski JV (1980) The obligatory role of the endothelial cells in
Plane et al. (1997) and Pratt et al. (1998). Although nifedipine
the relaxation of arterial smooth muscle by acetylcholine. Nature 288:373–376.
Hecker M, Bara AT, Bauersachs J and Busse R (1994) Characterisation of endothe-
reduced the response to anandamide as was reported for
lium-derived hyperpolarising factor as a cytochrome P450-derived arachidonic
bradykinin, charybdotoxin was without effect and, more con-
acid metabolite in mammals. J Physiol 481:407– 414.
Hu S and Kim HS (1993) Activation of Kϩ channels in vascular smooth muscle by
clusively, the cannabinoid receptor antagonist reduced the
cytochrome P450 metabolites of arachidonic acid. Eur J Pharmacol 230:215–221.
vasodilation to anandamide but was without effect on that to
Laurent F, Michel A, Chapat JP and Boucard M (1993) Evaluation of the relaxant
effects of SCA 40, a novel charybdotoxin-sensitive potassium channel opener in
bradykinin. Further, the time course of the vasodilator re-
guinea pig isolated trachealis. Br J Pharmacol 108:622– 626.
sponse to bradykinin and anandamide was dissimilar; that to
Ohlmann P, Martinez MC, Schneider F, Stoclet JC and Andriantsitohaina R (1997)
Characterization of endothelium-derived relaxing factors released by bradykinin
anandamide was slow in onset and of prolonged duration.
in human resistance arteries. Br J Pharmacol 121:657– 664.
Oyekan AO, McGiff JC, Rosencrantz-Weiss P and Quilley J (1994) Relaxant re-
Acknowledgment
sponses of rabbit aorta: influence of cytochrome P450 inhibitors. J Pharmacol Exp Ther 268:262–269.
The authors thank RBI for the gift of SR 171416A.
Plane F, Holland M, Waldron GJ, Garland CJ and Boyle JP (1997) Evidence that
anandamide and EDHF act via different mechanisms in rat isolated mesenteric
References
arteries. Br J Pharmacol 121:1509 –1511.
Popp R, Bauersachs J, Hecker M, Fleming I and Busse R (1996) A transferable,
Bauersachs J, Hecker M and Busse R (1994) Display of the characteristics of
-naphthoflavone-inducible, hyperpolarizing factor is synthesized by native and
endothelium-derived hyperpolarising factor by a cytochrome P450-derived arachi-
cultured porcine coronary endothelial cells. J Physiol 497:699 –709.
donic acid metabolite in the coronary microcirculation. Br J Pharmacol 113:1548 –
Pratt PF, Hillard CJ, Edgemond WS and Campbell WB (1998) N-arachidonyleth-
anolamide relxation of bovine coronary artery is not mediated by CB1 cannabinoid
Bornheim LM, Kim KY, Chen B and Correia MA (1993) The effect of cannabidiol on
receptor. Am J Physiol 274:H375–H381.
mouse hepatic microsomal cytochrome P450-dependent anandamide metabolism. Biochem Biophys Res Commun 197:740 –746.
Quilley J, McGiff JC and Fulton D (1997) Pharmacological evaluation of an epoxide
Campbell WB, Gebremedhin D, Pratt PF and Harder DR (1996) Identification of
(EET) as the putative mediator of cytochrome P450-dependent (P450) vasodilation
epoxyeicosatrienoic acids as endothelium-derived hyperpolarising factors. Circ Res
to bradykinin (BK) in the rat heart. Hypertension 29:888 Abstr. 78:415– 423.
Rapacon M, Mieyal P, McGiff JC, Fulton D and Quilley J (1996) Contribution of
Devane WA, Hanus I, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D,
calcium-activated potassiumchannels to the vasodilator effect of bradykinin in the
Mandelbaum A, Etinger A and Mechoulam R (1992) Isolation and structure of a
isolated, perfused kidney of the rat. Br J Pharmacol 118:1504 –1508.
brain constituent that binds to the cannabinoid receptor. Science 258:1946 –1949.
Randall MD, Alexander SPH, Bennett T, Boyd EA, Fry JR, Gardiner SM, Kemp PA,
Edwards G, Zygmunt PM, Hogestatt ED and Weston AH (1996) Effects of cyto-
McCulloch AI and Kendall DA (1996) An endogenous cannabinoid as an endothe-
chrome P450 inhibitors on potassium currents and mechanical activity in rat
lium-derived vasorelaxant. Biochem Biophys Res Commun 229:114 –120.
portal vein. Br J Pharmacol 119:691–701.
Taylor SG and Weston AH (1988) Endothelium-derived hyperpolarizing factor: a new
Fukao M, Hattori Y, Kanno M, Sakuma I and Kitabatake A (1997) Evidence against
endogenous inhibitor from the vascular endothelium. Trends Pharmacol Sci
a role of cytochrome P459-derived arachidonic acid metabolites in endothelium-
9:272–274.
dependent hyperpolarization by acetylcholine in rat isolated mesenteric artery.
White R and Hiley CR (1998) Studies on the effects of carrabinoid receptor ligands
Br J Pharmacol 120:439 – 446.
in the small mesenteric artery of the rat. Br J Pharmacol 123:65P.
Fulton D, McGiff JC and Quilley J (1992) Contribution of NO and cytochrome P450
Zygmunt PM, Edwards G, Weston AH, Davis SC and Hogestatt ED (1996) Effects of
to the vasodilator effect of bradykinin in the rat kidney. Br J Pharmacol 107:722–
cytochrome P450 inhibitors on EDHF-mediated relaxation in the rat hepatic
artery. Br J Pharmacol 118:1147–1152.
Fulton D, Mahboubi K, McGiff JC and Quilley J (1995) Cytochrome P450-dependent
effects of bradykinin in the rat heart. Br J Pharmacol 114:99 –102. Send reprint requests to: Dr. J. Quilley, Department of Cell Biology,
Fulton D, McGiff JC and Quilley J (1994) Role of Kϩ channels in the vasodilator
response to bradykinin in the rat heart. Br J Pharmacol 113:954 –958.
A 65-year-old male patient presented with progressive shortness of breath over the past 3 months, recently even at rest. He neither complained about fever nor about chest pain. The laboratory findings were normal with only a slight increase in leuco-cytes. A significant restrictive ventilation disorder was noted on plethysmography. His past medical history included three vessel coronary artery dis
Austral Yachts Clubman 36 Review Product Information Issue: March 2003 Manufacturer: Austral Yachts For a summer afternoon the marina seems exceptionally quiet as my mate David and I step it out along thepontoons trying not to miss our 3.30pm scheduled arrival time. We figure most people are still sleeping offtheir New Year’s hangovers, or catching up on jobs around home. I hope I’v
 Fulton and Quilley
Fulton and Quilley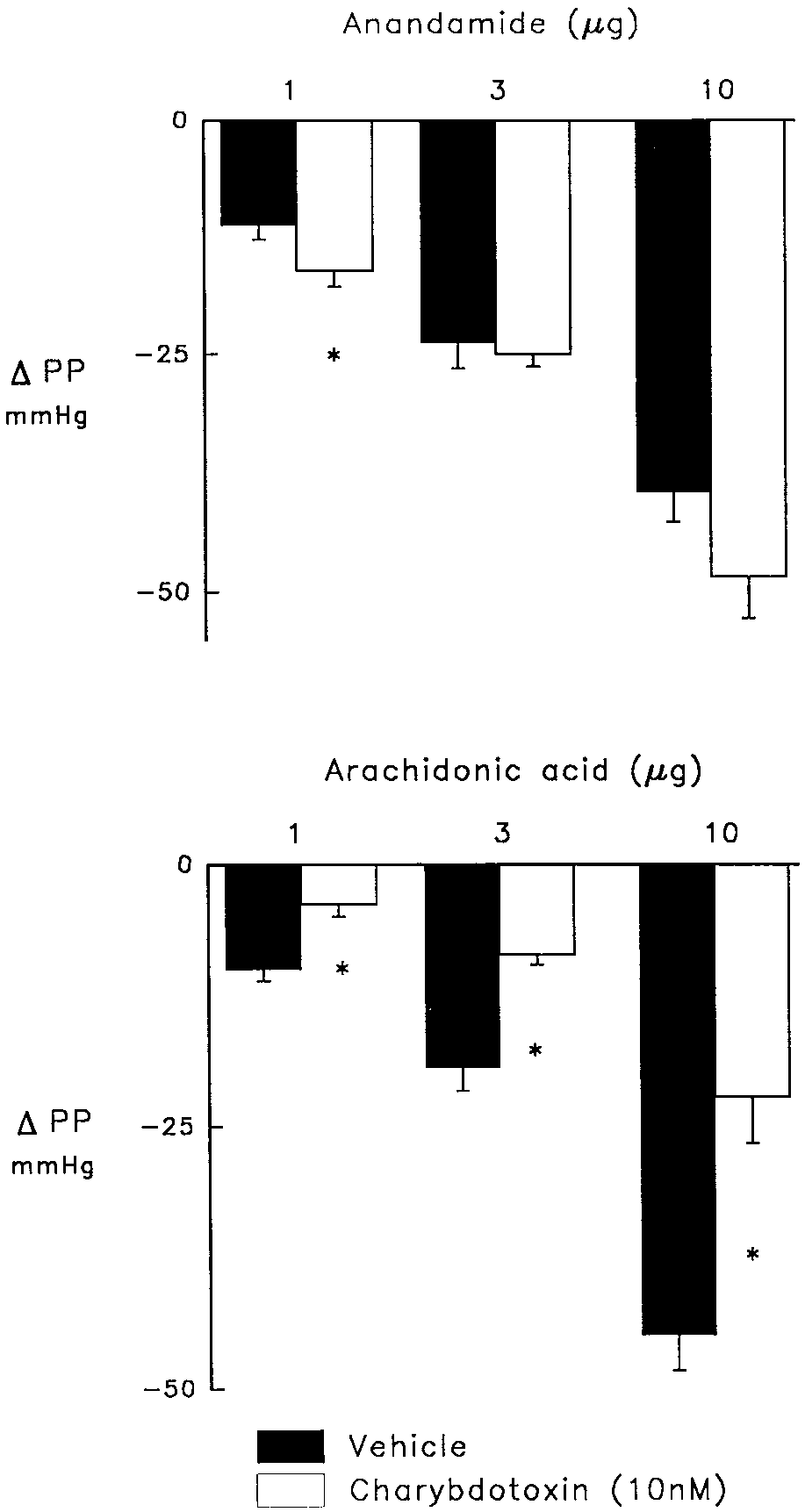
 Anandamide and Vasodilation to BK
Anandamide and Vasodilation to BK