Tadalafil appartiene alla classe degli inibitori selettivi della fosfodiesterasi di tipo 5, con un profilo farmacocinetico caratterizzato da un’emivita terminale di circa diciotto ore. Dopo somministrazione orale viene assorbito rapidamente e raggiunge concentrazioni plasmatiche massime in due ore. La biotrasformazione avviene principalmente tramite CYP3A4 con formazione di metaboliti inattivi, escreti in prevalenza con le feci. L’elevato legame alle proteine plasmatiche (>90%) assicura una distribuzione stabile. Nei confronti delle altre molecole della stessa classe, cialis compresse italia è noto per la durata prolungata dell’attività farmacologica.
Ismb
Analysis of Gene Expression Microarrays for Phenotype Classification
Andrea Califano , Gustavo Stolovitzky, Yuhai Tu
IBM Computational Biology Center, T.J. Watson Research Center, PO Box 704, Yorktown Heights, NY 10598
1 Introduction Recent advances in DNA microarray technology are Abstract
for the first time offering us exhaustive snapshots of some ofthe cell’s most intimate genetic mechanisms. These are cre-
Several microarray technologies that monitor the level of
ating a unique opportunity to improve our knowledge of the
expression of a large number of genes have recentlyemerged. Given DNA-microarray data for a set of cells
cellular machinery. Previous work on DNA microarrays has
characterized by a given phenotype and for a set of control
concentrated primarily on the identification of coregulated
cells, an important problem is to identify “patterns” of gene
genes to decipher the underlying structure of
expression that can be used to predict cell phenotype. The
genetic networks and/or the molecular classification of dis-
potential number of such patterns is exponential in the
In this paper, we propose a solution to this problem based
been proposed which aim at extracting useful information
from microarray data. Two general strategies have been fol-
substantially from previous schemes. It couples a complex,
lowed, based on either supervised or unsupervised
learning algorithms. In these approaches, the
probability of discovering discriminative gene expression
quantitative expression of n genes in k samples are consid-
patterns, and a pattern discovery algorithm called SPLASH. The latter discovers efficiently and deterministically all
ered as either n vectors in k-dimensional space or as k vec-
statistically significant gene expression patterns in the
tors in n-dimensional space. Various metrics measuring
phenotype set. Statistical significance is evaluated based on
distance between these vectors have been proposed.
the probability of a pattern to occur by chance in the control
In the unsupervised learning category, hierarchical clus-
set. Finally, a greedy set covering algorithm is used to
tering has been used to produce a hierarchical dendro-
select an optimal subset of statistically significant patterns,
gram in which genes with similar expression patterns
which form the basis for a standard likelihood ratio
(according to standard correlation coefficient metric) are
adjacent, and adjacency is interpreted as functional similar-
We analyze data from 60 human cancer cell lines using
ity. Other cluster analysis approaches attempt to
this method, and compare our results with those of other
make a partition of the n genes and/or the k samples into
supervised learning schemes. Different phenotypes are
homogeneous and well separated groups whose elements are
studied. These include cancer morphologies (such asmelanoma), molecular targets (such as mutations in the p53
interpreted as functionally related.
gene), and therapeutic targets related to the sensitivity to an
anticancer compounds. We also analyze a synthetic data set
designed to assess whether or not cells belong to a class
that shows that this technique is especially well suited for
characterized by a known phenotype, based on the cells’
the analysis of sub-phenotype mixtures.
gene expression profiles. This is also known as the cell phe-
For complex phenotypes, such as p53, our method
notype prediction problem. Typically, these algorithms are
produces an encouragingly low rate of false positives and
first trained on two example sets: a positive example set or
false negatives and seems to outperform the others. Similar
phenotype set, which contains data for cells characterized by
low rates are reported when predicting the efficacy of
a predefined phenotype, and a negative example set or con-
experimental anticancer compounds. This counts among
trol set, which includes data for cells that do not exhibit that
the first reported studies where drug efficacy has been
phenotype. In a vector of “marker genes,” or signature,
successfully predicted from large-scale expression dataanalysis.
is used for classification. Marker genes are selected based onthe discriminative power of their individual statistics. In
Keywords:
it is shown that Support Vector Machine (SVM) based algo-
Expression analysis. Gene Expression Patterns. Tissue
rithms outperform other standard learning algorithms such as
Classification. Phenotype Classification. Clustering.
decision trees, Parzen windows and Fisher’s linear discrimi-nant.
In this paper we introduce a novel supervised learning
algorithm for cell phenotype prediction. While our objec-
1. Phone: (914)784-7827. Fax: (914)784-6223.
tives are similar in spirit to those reported in
have been analyzed according to a variety of phenotypes.
and [15] our method differs on several counts. First and fore-
The term “cell phenotype,” is generally used in the literature
most, rather than relying on a unique best-fit model, which
to indicate a common property of a set of cells. For instance,
optimally discriminates between the phenotype and control
a cancer morphology, such as melanoma, is a typical pheno-
set, we use multiple, optimally discriminative models. As
type. More subtle phenotypes are also possible and useful. In
supported by our results, this improves the analysis of com-
for instance, we refer to a “p53 phenotype”
plex phenotypes over single model techniques, especially
which identifies cancer cells with mutations in the p53 onco-
when those phenotypes are mixtures of multiple, simpler
gene. Also, by measuring the drug concentration required to
sub-phenotypes at the molecular level. Also, as opposed to
inhibit by 50% the cell line growth, the so-called GI50, it is
our analysis selects genes based on their collective dis-
possible to define a drug-sensitivity phenotype. This can be
criminative power, rather than on their individual one. In this
used to divide cells in two groups: one with cells that are
latter sense our method can be though of as complementary
inhibited by low concentrations of the drug (i.e., that are
highly sensitive to it) and the other with cells that require
Our algorithm finds gene groups whose expression is
high concentrations (i.e., that are resistant to it). A classifica-
tightly clustered in a subset of the phenotype set and not
tion method can then be used to predict whether an unknown
tightly clustered in any subsets of the control set. (We shall
cell line is likely to be sensitive or resistant to a given drug.
rigorously define what we mean by tightly in
Some complex phenotypes, such as the p53-related one,
Among these groups, an optimal subset is then used for clas-
are likely to be mixtures of simpler unknown sub-pheno-
sification. Given a microarray with Ng genes, there are 2
types at the molecular level, each one characterized by a pos-
sibly independent pattern. Methods that rely on a single
model are likely to perform poorly with these complex cases,
First, we transform the gene expression axis using a
as truly there is no single model that describes the entire set.
gene-dependent non-linear metric. This improves the
This is verified in where a systematic compara-
chances of finding tight clusters in the phenotype set that are
tive analysis of the classification performance of several
unlikely to occur in the control set (Section 2.1).
algorithms, including and has been performed
Second, rather than exploring all potential gene groups
using a standard leave-one-out cross-validation scheme.
by brute-force, we use an efficient pattern discovery algo-
Results are analyzed based on the sum of false positive and
rithm called SPLASH This is discussed in
Each discovered pattern is defined by a subset of the genes
For simple phenotypes, such as a specific cancer mor-
and by the subset of the phenotype set over which these
phology, a unique model is sufficient. In this case, perfor-
mance is similar for all methods. In more complex cases
Third, we discard all patterns that are not be statistically
such as with the p53-related phenotype, where multiple
significant under a null hypothesis which takes
models clearly emerge, our technique outperforms the other
into account some statistical properties of the control set,
two. We have also performed tests on synthetic datasets that
except for the correlations in the expression of different
mimic the statistics of the human cancer cell lines. This is
genes. In practice, genes are correlated and some patterns
useful in determining the practical limits of the technique,
may be deemed statistically significant even though they are
whether promiscuous patterns have a negative impact on
not. We shall call these promiscuous patterns. Promiscuous
classification performance, and whether the technique over-
patterns, however, are easily identified (see discussion in
fits the data, a typical problem of supervised learning algo-
and do not create serious consequences in the
rithms with many degrees of freedom. Our method is shown
Fourth, an optimal set of patterns is chosen among the
Finally, the analysis of the sensitivity to the drug
statistically significant ones using a greedy set covering
Chlorambucil shows consistently good results for both
algorithm For typical cases, this set is small, consisting
our method and SVM. These are among the first results
of one to three patterns. The combined pattern set is used to
where drug efficacy is predicted based only on large-scale
build a multivariate probability density model. A “control”
gene expression data from microarrays. In general, results
probability density model is also built for each gene from the
clearly indicated that accurate and sensitive phenotype pre-
samples in the control set. A standard classification scheme
diction, from absolute gene expression levels is possible.
based on the ratio of the two probability densities is then
The implication of our study is twofold. It could validate
used. This scheme allows us to decide whether or not a pre-
the possibility of creating diagnostic tools for the classifica-
viously unseen cell belongs to the phenotype set.
tion and identification of several diseases. It could also help
We have applied our supervised learning scheme to the
devise new tools to predict which one, among a set of alter-
classification of 60 human cancer cell lines from data
native therapies, may have the highest chances of success
obtained with Affymetrix HU6800 GeneChips Cell lines
with a pathology linked to a specific cell phenotype. How-
ever, we must be extremely cautious in extrapolating fromthe current analysis. The samples we have used are from cell
lines rather than patient tissue. As a result, they could be
much more homogeneous than real tissue samples. Also,
since these are all cancer cells, the set statistics could be
quite skewed. Therefore, these results would not be immedi-ately useful as a diagnostic tool. However, we believe thispaper constitutes proof of concept that successful phenotypeprediction can be accomplished from microarray data usingpattern discovery. Given more statistically sound phenotype
and control sets, this approach could be used to discover
Control-based data normalization. The shaded area
multiple sets of marker genes both for diagnostic and thera-
between two points (a) measures the distance between them. 2 Methods
In the following sections we will (1) describe rigorously
the metric used for distance/similarity measure; (2) describe
the pattern discovery algorithm; (3) determine the probabil-
In this new variable, the corresponding probability den-
ity of patterns to arise spontaneously in the control set; (4)
sity Q g(v) for the control set is uniformly distributed and
show how patterns can form the basis of a supervised learn-
normalized in the interval [0,1]. In Fig. 1b, the probability
density is plotted together with the transformed values for
2.1 Metric Definition and Data Normalization u , u , u , and u . As expected, the Euclidean distance
In order to find useful gene expression clusters or pat-
is now smaller than that between u
terns, we must first define a metric in the space of gene
and u , which makes them much more likely candidates for
expression values to best distinguish the “signal” (phenotype
a cluster. Indeed, if v = f (u) and v′ = f (u′) are two
set) from the “noise” (control set). In other words, in design-
transformed expression values, we shall take the Euclidean
ing an appropriate metric for our problem, we wish to mini-
metric in ν as our measure of similarity, or distance, between
mize the probability of discovering patterns that would be
likely to occur in the negative example set.
Suppose that the expression level u of the g-th gene, in
D(u, u′) ≡ v – v′ =
the control set, is distributed according to a given probability
density P g(u) . This density is estimated empirically, by
The above equation is intuitively equivalent to the defini-
using a sum of Gaussian densities centered around each
tion given before. In other words, the distance between two
expression measurement in the control set. The standard
expression values is chosen to be equal to the integral of the
deviation is a function of u and it is also computed empiri-
gene expression probability density in the control set
cally from repeatability studies. A sufficient number of sam-
between these two values. Since, the number of measure-
ples is required to measure this density with a sufficient
ments in the control set falling between u and u′ is propor-
degree of accuracy. In Fig.1a a possible shape for P g is
tional to the integral in it follows that the more
plotted along with four expression values from hypothetical
measurements in the control set fall between two values, the
phenotype cells a, b, c, and d: u , u , u and u . Although
further apart they are in the new coordinate system and vice
that between u and u , the likelihood of getting the former
In the following sections, we will use the transformed
values by chance is higher because they are very close to the
gene expression space. One significant advantage is that,
maximum of the expression probability density. In other
since the probability density for all genes in the control set is
words, if we want to minimize the probability of finding ran-
uniformly distributed in the transformed space over the inter-
dom clusters in the control set, we must choose a metric such
val [0, 1] , it is now possible to analytically compute the sta-
tistics of the patterns discovered in the control set. Based on
u and u . A natural choice is to renormalize the expres-
that, we can assign a statistical significance to patterns dis-
sion axis so that the distance between two points on the new
axis is equal to the integral of the P g(u) in the previouscoordinate system. This is accomplished by defining a newvariable v obtained by transforming the original variable uwith the following (gene specific) non-linear transformation
2.2 Definition of Gene Expression Patterns
Given a gene vector G = {1, 3, 4} and an experiment
vector E = {1, 2, 4} , V V
0.1 0.2 0.5 0.7 0.5 Ne = 4 expression pattern. We
V
because the values in its 2nd and 3rd column are spread over
an interval greater than 0.05. The same pattern, π in
Expression
is ( δ = 0.1 )-valid but not maximal for the matrix of Fig. 2,
Fig. 2: Example of a gene expression Matrix: the result of a
because adding gene 2 to G, produces π which is still δ -
valid. Pattern π is maximal because adding any other gene
or experiment yields submatrices that are no longer
gene expression levels, the level for each gene being roughly
proportional to the concentration of the mRNA transcribed
( j = 2, k = 5 )-pattern.
from that particular gene in the cell. N
gene probes in the microarray. N is the number of microar-
2.3 The Pattern Discovery Algorithm
ray samples (i.e., experiments or cells). Thus, a set of DNA
Full details of the SPLASH algorithm are given in
microarray experiments is conveniently represented by an
In that paper, SPLASH was introduced as an algorithm to
N × N gene expression matrix V = {v } , where e is
discover patterns in strings, where all possible relative
the experiment index and g is the gene index. From the last
strings alignment are allowed. Also, a density constraint is
to be the transformed expression level
introduced to limit the impact of random matches occurring
according to of the g-th gene in the e-th sample.
over large distances on the string. For the equivalent associa-
If the set is the control set, then the transformed gene expres-
tion discovery problem, relevant in this context, the approach
will be approximately uniformly distributed.
is analogous as we can imagine each row in the matrix to be
Gene vector and Experiment vector: A list of gene
equivalent to a string. However, the strings are prealigned inthe present case. In addition, the density constraint criteria
introduced in is no longer meaningful here, as the first
1 ≤ g < g < … < g ≤ N is called a gene vector. A
and last genes are as likely to form patterns as two corre-
E = {e , …, e } , w i t h
sponding to contiguous matrix columns.
1 ≤ e < e < … < e ≤ N is an experiment vector.
Using the notation of the canonical seed set P has
-valid jk-patterns: Let V be a gene expression matrix,
a single pattern with no genes, all the rows, and an offset of 0
then a gene vector G and an experiment vector E
for each row. The histogram operator T
uniquely define a j × k submatrix V
simply sorting the values in each column and then selecting
V. Given δ > 0 , V
all subsets of continuous values that are δ -valid. Non maxi-
column is tightly clustered in an interval of size up to
mal subsets that are completely contained within another
δ . By this we mean that the maximum and the mini-
subset are removed. Each subset is a potential superpattern
mum value of each column must differ by less than δ.
of a maximal pattern. The enumerate operator T
The length of the experiment vector j is called the sup-
applied iteratively to create all possible maximal combina-tions of these superpatterns. As a results, all patterns that
port of the jk-pattern. Intuitively, if δ is small, each
exist in the data are generated hierarchically by combining
gene in a jk-pattern is expressed at approximately the
together smaller superpatterns, with fewer genes. Non maxi-
same level across all the experiments in the experiment
mal branches are eliminated at each iteration, as soon as their
vector. However, because these are transformed values,
corresponding superpattern arises. This contributes to the
the actual gene expression interval may be large. Maximal patterns: A δ -valid jk-pattern is maximal if the following two conditions hold: (1) it cannot be 2.4 Statistical significance of Patterns in Gene
extended into a δ -valid jk’-pattern, with k′ > k , by
Expression Matrices When gene expression values are organized in a gene expres-
adding genes to its gene vector, and (2) it cannot be
sion matrix, jk-patterns may occur for any given value of δ .
extended into a δ -valid j’k-pattern, with j′ > j, by add-
Can any of these patterns occur merely by chance? In this
ing experiments to its experiment vector.
subsection we address this question by studying the statistics
Example. Consider the gene expression matrix V of
of patterns in any N × N matrix, whose elements are sta-
tistically independent of each other and have the same proba-
Fig. 3: (see matrix in Fig.
to the gene vector of amaximal pattern, e.g., gene
Gene Vector {1; 3; 4} {1; 2; 3; 4} {1; 2; 3; 4; 5}
bility distribution as that of the control set in the transformed
, N ) ≈ ζk[1 – ζ]Ng [1 (1 + j )kδk
An important observation is in order at this junction: we
do not mean that the expression values of different genes in“real-life” gene-expression matrices are independent random
variables. Rather, we intend to use such a model as the null
hypothesis of our statistical framework precisely to identify
terns in V is
any skew or co-regulation in the phenotype set. This null
hypothesis definition is based on two assumptions: (a) that
the probability densities for the expression levels of each
gene are the same as in the control set, and (b) that the gene
N is the total number of ways in which one can choose a
expression levels in different experiments and/or those of
gene and experiment vector. In we make an
different genes are independently distributed. When discov-
approximation that is valid when δ is small. This is consis-
ering patterns in the phenotype set, the statistical relevant
tent with the values used in the experimental section, typi-
patterns will be those for which the null hypothesis is
rejected. These are patterns whose constituent genes are
To verify the validity of and also to show
either distributed differently in the phenotype set than in the
as a function of j and k, we have per-
control set, and/or are expressed in a correlated fashion. Both
formed simulations by running pattern discovery on syn-
of these features are actually the kind of behavior that we are
thetic gene expression matrices, using the statistics of the
seeking to differentiate the two sets.
null hypothesis. shows an excellent agreement
Of course, many genes are not independently distributed
between various theoretical and experimental values of N
in the control set. Therefore patterns may arise that reject the
null hypothesis and yet are likely to occur in the control set.
ment is observed for other value of the parameters. In
We shall call these promiscuous patterns. Promiscuous pat-
terns, are easily eliminated in a post-processing phase and do
≈ N [ jδj 1 ( j – 1)δj
not contribute significantly to remaining analysis. This is
From the analysis of the variance of the number of jk-pat-
verified by experimental results in where any
terns (not reported here) we have also verified that this is
correlation in the control set is artificially removed. Results
typically very close to the mean number of jk-patterns, espe-
for this set are not different from those on the real data,
cially when the latter is small. This result suggests that the
showing that gene correlation in the control set is not an
distribution of the number of patterns could be well approxi-
mated by a Poisson distribution. Indeed the histogram of the
Our main result on the statistics of patterns is the follow-
number ν of jk-patterns is very well fitted by the distribution
ing: given δ > 0 , an N × N
gene expression matrix V, a N e N jk/ν! (data not shown). k-dimensional gene vector G and a j-dimensional experiment
We can use the previous observations to assess the statis-
vector E, the probability that the submatrix V
tical significance of a pattern in the phenotype set with
respect to the randomized control set. Using classical statis-tics reasoning, we reject maximal δ -valid jk-patterns thatwould be likely to occur in the randomized control set. Under the null hypothesis, the probability p
more jk-patterns occur in the phenotype set is
1. The derivation of this formula is lengthy and will be pub-
lished elsewhere. The interested reader can request a draft
This will be the p-value or significance level of our statistical
test. Thus, setting a reasonable threshold P , we can say that
Fig. 4: Average number of δ -valid jk-patterns in randomly generated synthetic data. Continuous curves are the theoretical values. Dots are
experimental values obtained with Splash. N
if we observe one or more jk-patterns in the phenotype set
< P , we reject the null hypothesis and conjecture
that such jk-patterns could be specific of the cell phenotype
Using this score, we can easily determine whether promiscu-
2.5 Classification
ous patterns are contained in the set of statistically signifi-
Once the statistically significant patterns are found in the
cant patterns. Patterns with positive values of S for samples
phenotype set, we can use them as classifiers to build a dis-
taken from the control set are considered promiscuous. Next
criminant function. This function should determine whether
we assign the statistically significant patterns a promiscuity
or not a previously unseen sample v = (v , …, v
belongs to the phenotype or the control set. To this end we
S (v ∈ Control Set) ,
build a model for the probability density function of the
S (v) > 0
expression level for each statistically significant pattern π
of the phenotype set. Each i-th gene of π contributes with a
where the sum runs over all the samples in the control set for
factor P (v) . The probability density P is chosen to be
which S > 0 . Patterns whose S < 0 for all samples in the
normally distributed with mean equal to the average of the
control set have a promiscuity index of zero. Patterns can
cluster for the i-th gene and standard deviation σ = δ ⁄ 2 .
now be sorted according to the promiscuity index, with the
This method is preferred over an empirical derivation from
actual measurements because patterns are typically sup-
The next step is to associate each pattern π with a cov-
erage set, which includes all the samples in the phenotype set
For samples in the control set, the same gene would be
with a positive score S (v
expressed according to a different probability density
Finally, an optimal set of patterns is selected using a
P (v) . The latter can be built empirically because all the
greedy set covering algorithm to optimally cover the
samples in the control set can be used. As discussed earlier,
phenotype set. The set covering algorithm tries to use the
we use a sum of Gaussian densities with a mean equal to the
patterns in sort order according to the promiscuity index: the
value of the sample measurement and a standard deviation
least promiscuous and most covering pattern is chosen first.
derived from repeatability experiments.
The smallest subset of patterns whose coverage sets opti-
On a first order approximation, we shall assume indepen-
mally cover the phenotype set is then used for classification
dence between genes and take the multivariate distribution to
purposes. Therefore, if a non-promiscuous set that optimally
be equal to the product of the probability densities of the
covers the phenotype set exists, it will be selected over a pro-
individual genes, both in the phenotype and in the control
set. This assumption is necessary because we do not have
enough data to construct a realistic multidimensional proba-
ranges between one and three. The score of a previously
unclassified sample v is defined as
Promiscuous patterns, which arise from correlations in
S(v) = max(S (v), l = 1, …, N ) .
the control set, are likely to play a minor role in the classifi-cation as described in the following discussion. To determine
Given a threshold S , the sample fits the phenotype model
if a new microarray sample fits the phenotype model of a jk-S . The theoretical false positive (FP) and
pattern π for the expression values (v , v , …, v ) over
false negative (FN) probabilities can be easily estimated by
the k genes that constitute π , we score it by the logarithm of
and P over the region where their ratio is
greater or smaller than the threshold. If a single classifier isused, S
= 0 minimizes the sum of false positive and false
negative probabilities In the multivariate model, ScFig. 5: Histogram of the log expression of gene 135.
expression of the nonbasal activity for all genes.
must be tuned. S is an useful tunable parameter practically
since different problem requires different balance between
3 Results
An Affymetrix HU6800 GeneChip has been used to
monitor the gene expression levels of 6,817 full length
human genes in 60 human cancer cell lines These are
organized into a set of panels for leukemia, melanoma, and
In H ( x) is the Heaviside function,
cancer of the lung, colon, kidney, ovary, and central nervous
( H ( x ≥ 0) = 1 and H ( x < 0) = 0 ), which results from
system. The identity of the genes is not known to the authors.
the integration of the delta function in the u distribution.
They are therefore identified by a numeric id.
For α ≠ 0 , different values of u can correspond to the
Genes with expression values of 20 or less are considered
same value of v . This is not a problem unless α is of the
switched off. From the 6,817 original genes, a subset of 418
same order of δ , in which case we discard the values at u
was selected by means of a variational filter to eliminate
genes that did not change significantly across samples (varia-
3.1 Phenotype Analysis
The fluorescence intensity φ of each gene, roughly pro-
Given a gene expression matrix V, with renormalized values
portional to the mRNA concentration, appears to be lognor-
, the pattern discovery algorithm SPLASH is used to find
mally distributed. The value of variable u is then chosen as
all maximal δ -valid jk-patterns for k ≥ 4 and j ≥ 4 . These
u = log (φ ) . In the histogram of a typical gene’s
parameters are chosen because patterns with too few genes
expression over the 60 samples is shown. This distribution is
are not specific enough, while patterns with too small a sup-
clearly bimodal. There is a peak at the basal level, corre-
port do not characterize a significant consensus in the
sponding to the gene being switched off in some experi-
dataset. δ is chosen between 0.05 and 0.15, depending on
ments. The non-basal expression values, on the other hand,
the dataset, such that a sufficient number of patterns is dis-
are distributed with a well behaved mean and standard devia-
covered, typically on the order of 10 to 50 statistically signif-
tion. Thus, we write the corresponding probability density
icant patterns. Larger values of δ are possible but they
increase the probability of finding promiscuous pattern and
reduce performance. In general, the smaller value of δ one
P (u ) = α δ(u – u ) + (1 – α )P
can choose and still discover patterns, the better the results.
where α is the percentage of expression data being at the
The threshold of significance P is chosen to be 10
(u ) is the density function for the
We discuss experimental results on the classification of 7
non-basal expression values. For each gene, we determine its
samples in the melanoma panel, of 17 samples with muta-
basal level α , the mean u , and standard deviation σ of its
tions in the p53 gene, and of 10 samples whose growth is
(u ) . Non-basal values of u are nor-
highly inhibited by the drug Chlorambucil. For each experi-
mally distributed in accord with the observed lognormal dis-
tribution of the φ .This is shown in where we plot the
and false negative probabilities as a function of the matching
combined distribution obtained by shifting and rescaling the
nonbasal activity of each gene as u′ = (u – u ) ⁄ σ
Three methods are studied: our pattern discovery method
(PD), the support vector machine (SVM) method of [15];and the gene by gene method (GBG) of [11]. For each givenphenotype, we use its complement in the NCI-60, excluding
Fig. 7: Classification performance for (a) Melanoma, (b) Complex Synthetic phenotype, (c) p53 and (d) Chlorambucil. The sum of the false positive and false negative probabilities is plotted as a function of the classification score threshold. PD is shown by a thick Solid line, SVM by a dashed line with diamonds, and GBG by a dotted line with circles.
the samples whose phenotype cannot be accurately deter-
significant range of the match threshold S where both false
mined (neutral samples), as the control set.
positive and false negative probabilities are zero. This is con-
Given the limited number of samples in the NCI-60 set,
sidered perfect recognition. The GBG produces results
false positive and false negative ratios are computed by cross
which are very similar, although a fraction less accurate. The
validation. Each sample both in the phenotype and in the
time required to classify a sample with the PD method is
control set is removed in turn. The algorithm is trained using
the remaining samples, this includes gene axis transforma-
p53 Mutation: A more challenging phenotype is that of 17
tion, pattern discovery, and set covering. Finally, the previ-
samples for cells with mutations in the p53 gene. The corre-
ously removed sample is classified as described in
sponding set of cancer morphologies is considerably more
When a phenotype set sample is misclassified it is con-
complex. It includes 5 melanoma, 3 renal cancer samples, 2
sidered a false negative. When a control sample is misclassi-
samples for cancer of the central nervous system, leukemia,
fied it is considered a false positive. All computation times
ovarian cancer, and breast cancer, and 1 sample for colon
reported are relative to a 450MHZ Pentium II.
cancer. As mentioned earlier, this is likely to have several
Melanoma: The melanoma panel includes 7 samples. There
sub-phenotypes at the molecular level. This is confirmed by
are also 14 neutral samples. These have been selected by
our analysis, which also highlights a much wider range of
biologists prior to this analysis. When the complete set of
variability for the various methods. As shown in the
melanoma samples is used for the training, there is only one
statistically significant gene expression pattern that is
selected after the set covering phase. a shows the per-
result, bringing that value to about 0.46. Our pattern discov-
formance of the complete analysis, with δ = 0.12 , as
ery based approach, with δ = 0.12 , has the best result at
described in the previous section. Both SVM and PD show a
) = 0.33 . Three distinct, rather orthogonal pat-
same gene by gene statistics as the control set for the p53
terns are used on average for each sample classification. If
study. A set of 48 control samples have been generated from
only one pattern is allowed, results become close to that of
this model at random. Their gene by gene probability density
the SVM method. The time required to classify a sample
is virtually identical to that of the real control set in the p53
with the PD method is approximately 20 seconds.
study. A set of 18 phenotype samples has been synthetically
Chlorambucil GI50: Some truly interesting phenotypes are
generated. This set consist of three independent sub-pheno-
associated to the ability of a given drug to inhibit cell
types of 6 samples each. Each sub-phenotype is character-
growth. These are relevant because many experimental anti-
ized by 10 marker genes clustered around a tight interval.
cancer compounds exhibit relatively poor growth inhibition
Remaining genes are modeled as in the control set. Marker
rates across large variety of cancer cells of similar morphol-
genes are different for different sub-phenotype with some
ogy. If one could however correlate the effectiveness of a
overlap. In particular, sub-phenotype 1 and 2 have 6 marker
compound to the much richer space of the gene expression
genes in common; sub-phenotype 2 and 3 have 2 marker
profile of a cell, it could be possible to determine a-priori
genes in common. Marker genes are expressed differently
which cells are most likely to be inhibited by a drug. To test
than in the control according to the following criteria: 1) they
this scenario, we have selected Chlorambucil (NSC 3088)
have a different mean located about 0.5σ away from the
from the NCI anti-cancer database. Since the growth inhibi-
control mean, 2) they have a smaller standard deviation
tion rate is distributed rather continuously over the entire
0.33σ , where σ is the standard deviation of the same gene
NCI-60 spectrum, we have split the samples in three groups.
in the control set. As shown in there is a dramatic dif-
The phenotype group, contains the 10 cells that are most
ference between the performance of the SVM method and
inhibited by Chlorambucil. The control group contains the
that of the PD method. The minimum of p
20 samples whose growth is least inhibited by the com-
for the SVM method and 0.19 for the PD method. About
pound. The third set of 30 cells is considered neutral. As
80% of the genes composing the sub-phenotypes are cor-
shown in the SVM and PD methods perform simi-
rectly identified by the 3 resulting patterns. Of course, we
larly, with a slight advantage towards the former. Best values
must be careful not to draw conclusions from this simple
are 0.35 and 0.4 respectively. For the PD method, a
minded exercise. Yet, this seems a good indication that the
value of δ = 0.12 is used. The other method cannot to bet-
PD method is a suitable choice for the classification of some
ter than 0.55. The time required by the PD method to classify
complex phenotypes that may be mixtures of simpler sub-
The second synthetic data set has been designed to deter-
3.2 Synthetic data analysis
mine whether correlation of genes in the control set is a sig-
Several cross-validation checks using synthetic or random-
nificant factor and could reduce the performance of the
ized data have been performed to validate our approach.
technique. To accomplish this goal, the values of the genes
Three synthetic data sets are analyzed.
have been randomly permuted only across the control set on
The first test has been designed to evaluate the theoretical
a gene by gene basis. This has the effect of leaving the
performance of the algorithms in the case of phenotype mix-
expression probability density for each gene for the control
tures. A synthetic data model has been generated with the
virtually unchanged, while removing any possible correla-tion between the values of
Results for the classification are shown in (squares).
There are no major differences with respect to the same
curve for the real data of and reproduced in as
the thick solid line. This proves that correlation in the geneseven though present in the data does not result in classifiers
that are highly correlated over the control set.
Finally, we have designed a test to determine whether
this approach may suffer from overfitting the data. To thatend, classification has been performed using the same data
and criteria as for the p53 phenotype study but after the
expression values of the individual genes have been ran-domly permuted across all the samples on a gene by gene
basis. The results of the classification are shown as triangles
in As clearly shown, performance is very poor, with a
consistently larger than 1 over the entire clas-
Fig. 8: Performance of the PD method with the actual p53 training
sification threshold interval. The PD method, as well as the
randomized control and phenotype sets (triangles).
SVM and GBG methods, exhibits no predictability for these
data sets, i.e., the sum of the false positive and the false neg-
Hart, Laxmi Parida, and Ruud Bolle for many useful discus-
ative rates is close to 1, as it should be.
sions on pattern discovery and statistics, Donna Slonim for acareful reading of the manuscript and Hilary Coller for very
4 Summary and Conclusions
helpful insights on the NCI-60 data.
Based on a combinatorial multivariate approach, we have
developed a systematic framework for cell phenotype classi-
6 References
fication from gene expression microarray data. We have used
Lockhart, D. J.; Dong, H.; et al. (1996). Expression
SPLASH, a deterministic pattern discovery algorithm, to dis-
monitoring by hybridization to high-density oligonu-
cover all gene expression patterns on a given data set.
cleotide arrays. Nature Biotechnol. 14:1675-1680.
We have then evaluated analytically the statistical signifi-
Brown, P.O.; and Botstein, D. (1999). Exploring thenew world of the genome with DNA microarrays,
cance the patterns. The set of statistically significant patterns
form the basis our classification scheme. A scoring system,
DeRisi, J.; Penland, L; et al. (1996). Use of a cDNA
based on the ratio of the probabilities between the phenotype
microarray to analyse gene expression patterns in
and the control sets is constructed in accordance with the
human cancer. Nature Genetics 14:457-460.
multivariate nature of the patterns.
Wodicka, L.; Dong, H.; et al. (1997). Genome-wide
We have applied our classification method to gene
expression monitoring in Saccharomyces cerevisiae.
expression data from 60 human cancer cell lines. Results for
the classification of melanoma, p53 mutations and the GI
Cho, R. J.; Campbell, M. J.; et al. (1998). A genome-
wide transcriptional analysis of the mitotic cell cycle.
activity of Chlorambucil are excellent. They range from 0%
to about 40% sum of false positive and false negative proba-
Chu, S.; DeRisi,J.L.; et al. (1998). The transcriptional
bility. The high sensitivity and specificity of the method for
program of sporulation in budding yeast. Science
complex phenotypes such as p53 show that the method can
successfully deal with multiple independent sub-phenotypes.
Iyer, V. R.; Eisen, M.B.; et al. (1999). The transcrip-
We also report results from one of the first attempts to pre-
tional program in the response of human fibroblasts to
dict drug effectiveness from gene expression data.
We compare our method with other supervised learning
DeRisi, J.L.; Iyer, V.R.; et al. (1997). Exploring themetabolic and genetic control of gene expression on a
schemes: the gene by gene method of and the support
vector machine method of The classification based on
Perou, Ch.; Jeffrey, S.S.; et al. (1999). Distinctive gene
pattern discovery performed almost as good or better than
expression patterns in human mammary epithelial cells
the other methods. However these comparisons depend
and breast cancers, Proc Natl Acad Sci U S A 96:9212-
strongly on the data set under classification. One relevant
conclusion in this context is that our approach is especially
[10] Alon, U.; Barkai, N.; Notterman, D.A.; Grish, K.; Yba-
well suited to treat complex phenotypes, composed of sev-
rra, S.; Mack, D.; and Levine, A.J. (1999). Broad pat-terns of gene expression revealed by clustering analysis
eral sub-phenotypes. We showed that such is the case for the
of tumor and normal colon tissues probed by oligonu-
p53-related phenotype and for a synthetic data set.
cleotide arrays. Proc Natl Acad Sci U S A 96:6745-
Besides the better predictive power, another advantage of
our method over SVM is that our method highlights the rele-
[11] Golub, T.R.; Slonim, D.K. ; et al. (1999), Molecular
vant marker genes, their expression range in the phenotype,
classification of cancer: class discovery and class pre-
and the independent patterns that relate them. Such informa-
diction by gene expression monitoring. Science 286:531-537.
tion is highly desirable for discovering the mechanism for
[12] Eisen, M. B., Spellman, P. T.; et al. (1998). Cluster
various diseases at the molecular level. The SVM method, on
analysis and display of genome-wide expression pat-
the other hand, is more like a black box for classification.
terns. Proc Natl Acad Sci U S A 95:14863-14868.
The method described in this paper is a significant contri-
[13] Tamayo, P., Slonim, D.; et al. (1999). Interpreting pat-
bution to the set of tools for the analysis of gene expression
terns of gene expression with self-organizing maps:
microarray data. It constitutes proof of concept for a range of
methods and application to hematopoietic differentia-
important practical applications for diagnostics and for the
tion. Proc Natl Acad Sci U S A 96: 2907-2912.
design of highly specific therapies. It may also help under-
[14] Ben-Dor A and Yakhini Z. (1999). Clustering Gene
Expression Patterns. In Proc. of the 3rd International
standing the structure of the underlying gene regulatory net-
Conference on Computational Molecular Biology, 33-
works and ultimately the mechanism responsible for various
[15] Brown, M.P.S.; Grundy, W.N.; Lin, D.; Cristianini, N.;
Sugnet, C.; Ares, M.; and Haussler, D. (1999). Support
5 Acknowledgments
Vector Machine Classification of Microarray Gene
Special thanks go to the Whitehead Institute team, Jill
Expression Data, University of California Technical
Mesirov, Donna Slonim, and Pablo Tamayo for the Gene
Report USCC-CRL-99-09. Available at: http://
Expression datasets. We also thank Ajay Royyuru, Reece
www.cse.ucsc.edu/research/compbio/genex. [16] D’haeseller, P.; Wen X.; Fuhrman, S.; and Somogyi, R.
(1998). Mining the Gene Expression Matrix: InferringGene Relationships from Large Scale Gene ExpressionData. In Information Processes in Cells and Tissues,203-212, Paton, R.C. and Holcombe, M. Eds., PlenumPublishing. [17] Califano, A. (1999), SPLASH: Structural Pattern
Localization Algorithm by Sequential Histograming. Bioinformatics, in press. Preprints available at http://www.research.ibm.com/topics/popups/deep/math/html/splashexternal.PDF. [18] Chvatal, V. (1979) . A greedy heuristics for the set cov-
ering problem. Math. Oper. Res. 4:233-235. [19] Welch, B.L. (1939). Note on Discriminant Functions. [20] Lehmann, E.L. (1986). Testing Statistical Hypotheses. [21] Weinstein, J. N.; Myers, T. G.; et al. (1997). An infor-
mation-intensive approach to the molecular pharmacol-ogy of cancer. Science 275:343-349. [22] Various activity data for Chlorambucil on the NCI-60
cell lines can be found by using NSC access number3088 on site http://dtp.nci.nih.gov.
Fig. 9: Normalized probability density: log- expression of the nonbasal activity for all genes.
Name __________________________________________________ Date ____________ Date of birth _____________________ Date of last eye exam ______________________Referring Dr. ______________________________ Primary Care Physician __________________________What is the chief complaint regarding your eyes?_______________________________________________________________________________________________
Dr. Woods Knee Surgery Discharge Instructions WOUND CARE There may be a slight bruising along the wound with discoloration. This bruising may also move down into your calf ankle and foot. There may be a small amount of drainage from the incision area. ___ Keep dressing clean, dry and in place until follow up appointment. ___ Remove dressing: ___ days, ___ physical therapy will remov
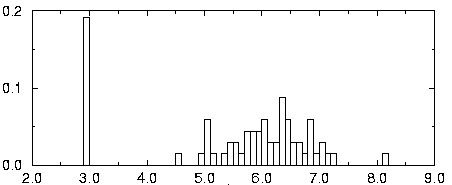 Fig. 5: Histogram of the log expression of gene 135.
Fig. 5: Histogram of the log expression of gene 135.